- Detalles
- Categoría: Comsol
- Visto: 8324
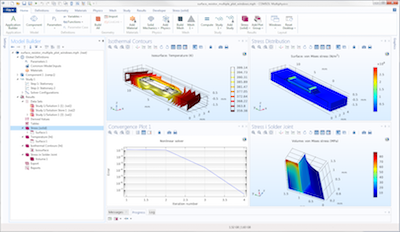
COMSOL Desktop® es el entorno de usuario integrado para crear y administrar simulaciones en el programa COMSOL Multiphysics®. Incluye la ventana Model Builder o Constructor del modelo, el Árbol del modelo o Model Tree, la ventana de Gráficos, así como los menús y las barras de herramientas con una multitud de herramientas de modelado. En esta publicación del blog de COMSOL, Magnus Ringh nos muestra cómo personalizar la interfaz de usuario y utilizar una variedad de atajos de teclado para aprovechar al máximo el proceso de modelado.
Entre los trucos que nos explica, nos muestra cómo maximizar la ventana de gráficos y restablecerla para que el entorno vuelva a estar presente; cómo cambiar el tamaño de las ventanas, ocultarlas, ponerlas flotantes o cerrarlas; organizar las ventanas gráficas para mostrar múltiples gráficos; restaurar el escritorio a un diseño predeterminado; realizar la personalización en los entornos Linux® y macOS; y conocer los atajos de teclado más útiles para el modelado con COMSOL Multiphysics®.
- Detalles
- Categoría: Maple
- Visto: 5271

Destacados
|
Está bien, lo conseguimos. Sabemos que ama Maple, pero algunos años se pregunta si vale la pena actualizar. La gente usa Maple para hacer muchas cosas diferentes, por lo que es inevitable que cada nueva versión incluya algunas características que le interesen y otras que realmente no necesite.
Pero si aún no está utilizando Maple 2018, realmente se lo estás perdiendo. Tal vez se pierde más de lo que se cree.
Para ayudarle a tomar una decisión, este documento técnico describe algunas de las novedades de Maple en cada uno de los últimos tres lanzamientos. Una vez que haya tenido la oportunidad de aprender acerca de lo que ha pasado en Maple desde la última actualización, pregúntese: ¿hay realmente algo de Maple 2018 que no quiera utilizar?
- Detalles
- Categoría: Comsol
- Visto: 19501

En esta interesante entrada del blog de COMSOL, Ed Fontes nos expica cómo algunas ideas sobre la bondad de los métodos de resolución de problemas de mecánica de fluidos (CFD) muchas veces vienen dadas por hechos poco fundados. Hablamos de la contraposición del método de los elementos finitos respecto al método del volumen finito.
Algunos ingenieros creen en la superioridad de los métodos de volumen finito en comparación con los métodos de elementos finitos. Pero según Ed, en general, no existe una verdadera base científica para esta opinión y en este artículo nos desgrana por qué diferentes métodos pueden ser adecuados para diferentes problemas.
Los métodos de elementos finitos son ampliamente utilizados por la comunidad de análisis numérico para estudiar métodos numéricos para el flujo de fluidos. Existe una gran cantidad de trabajos descritos en publicaciones científicas sobre CFD y métodos de elementos finitos, así como trabajos más recientes para los métodos nodales discontinuos de Galerkin (DG), que son métodos de elementos finitos con funciones de base discontinua.
Los paquetes comerciales para CFD se basan tradicionalmente en métodos de volumen finito. Esto se debe al hecho de que, básicamente, todos los paquetes comerciales más grandes para CFD tienen los mismos antepasados. Hay una gran cantidad de trabajo y tecnología invertidos en estos métodos en la industria. Se han implementado diferentes métodos para que puedan calcular e integrar los flujos de manera eficiente y precisa tanto para mallas estructuradas (por ejemplo, hexaedros) y no estructuradas (por ejemplo, tetraedros).
Dicho esto, no hay base teórica o práctica que sustente la hipótesis de que los métodos de volumen finito son superiores a los métodos de elementos finitos para el flujo de fluidos. Primero, hay muchos métodos diferentes de volúmenes finitos y métodos de elementos finitos y algunos de estos métodos se superponen. Segundo, la implementación de un método tiene un impacto muy grande en el uso práctico de un software. Podemos decir que el paquete X parece hacer un mejor trabajo que el paquete Y para una determinada familia de problemas. Sin embargo, esto no es porque uno usa FEM y el otro FVM.
Lea el artículo completo en el blog de COMSOL para entender la diferencia entre ambos métodos.
- Detalles
- Categoría: Minitab
- Visto: 13855
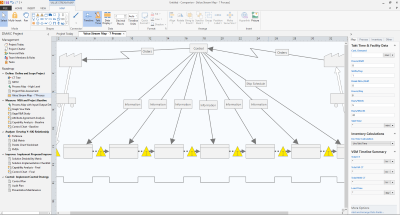
El mapeo de flujo de valor en una piedra angular de la metodología de mejora de procesos Lean, y también es una reconocida herramienta utilizada en Six Sigma. Un mapa de flujo de valor ilustra el flujo de materiales e información a medida que un producto o servicio se mueve a través de un proceso. Crear un mapa de flujo de valor del "estado actual" puede ayudar a identificar el desperdicio y también facilita la visualización de un estado mejorado para el proceso en el futuro.
Se puede utilizar el mapeo de flujo de valor para mejorar cualquier proceso. Pero a menos que se utilice la herramienta correctamente, el mapa de flujo de valor podría no capturar todas las oportunidades que se tienen para mejorar la calidad y la eficiencia.
Aquí se exponen cinco pautas para obtener el mayor beneficio de la energía puesta en el mapeo de flujo de valor:
- Capturar el proceso tal como funciona ahora, no como se supone que opera.
Un proceso que funcionó bien cuando se tenían 20 empleados puede no funcionar eficientemente ahora que la empresa es una compañía de 200 personas. Asegurarse de mapear el proceso tal y como ocurre ahora, no de la forma que solía funcionar, ¡o cómo le gustaría que funcionara! - Asignar un administrador del Mapa de Flujo de Valor para dirigir el esfuerzo del mapeo.
La aportación de los miembros del equipo y las partes es importante, pero se debe designar (o elegir) a un miembro del equipo para que dibuje el mapa completo de la cadena de valor. Esto asegura que el administrador entienda los flujos de material e información. - Recorrer el proceso para asegurarse de que el flujo de materiales e información son precisos.
Asegurarse de que el mapa refleje la realidad del proceso; verificarlo siguiendo el proceso desde el principio hasta el final puede revelar detalles cruciales que se podrían haber pasado por alto. - Concéntrarse en un pequeño paso cada vez. Asegurarse de capturar cada paso con precisión. Por ejemplo, no se debe confiar en el reloj de la pared para medir los tiempos de ciclo; debe utilizarse un cronómetro.
La creación de un mapa de flujo de valor del estado actual del proceso ayuda a concentrarse en áreas de desperdicio como el exceso de inventario, el tiempo sin valor añadido y múltiples operadores. A medida que se visualiza el estado futuro del proceso, pueden variarse datos en el mapa de estado actual para explorar los efectos de posibles mejoras.
Si bien es posible realizar el mapeo de flujo de valor en papel, las herramientas de software como las de Companion by Minitab hace que el mapeo de flujo de valor sea mucho más fácil.
- Detalles
- Categoría: BIOVIA
- Visto: 11178

Los ingenieros de hoy se encuentran en un aprieto, los requisitos de productos cada vez más avanzados están superando las capacidades de los materiales que están inmediatamente disponibles. Los componentes eléctricos deben ser más pequeños, los plásticos requieren materias primas más variadas y sostenibles, los edificios deben usar menos energía, los médicos desean utilizar dispositivos médicos adaptados paciente por paciente. Como resultado, los ingenieros deben encontrar soluciones cada vez más complicadas.
Este desafío se extiende más allá de qué material o combinación de materiales podría usar un ingeniero, las estructuras de los materiales o cómo se fabrica la pieza terminada pueden afectar el diseño final. Para aprovechar realmente los beneficios que ofrecen los materiales avanzados, los ingenieros deben adoptar un enfoque “multiescala” para el diseño de materiales, considerando cómo las diferentes escalas de longitud (es decir, desde macroscópicas a microscópicas a cuánticas) y diversas estructuras contribuyen a las propiedades generales de sus productos finales.
Las propiedades de los materiales son intrínsecamente multiescala. En pocas palabras, las características establecidas en el nivel cuántico se suman a través de los átomos, las moléculas y las redes (o la falta de ellas) para producir las propiedades que vemos a nivel global.
Sin embargo, no existe una fórmula única que pueda describir estos cambios a medida que avanzamos de la subatómica a la macroescala. Requiere construir puentes entre múltiples campos de la física y tener en cuenta los efectos aditivos de pequeños cambios en la pureza y estructura del material. Lograr este objetivo significa ir más allá de la experimentación física para utilizar los avances en el modelado y simulación de materiales. Existen herramientas que permiten a los científicos ejecutar y repetir rápidamente experimentos en silico para analizar más rápidamente diferentes combinaciones de materiales y estructuras para optimizar el rendimiento del producto final.
La adopción de herramientas de modelado y simulación, como Materials Studio, para aumentar el diseño del producto presenta una oportunidad única para los ingenieros: en lugar de diseñar una pieza según las restricciones de los materiales disponibles, pueden diseñar un material basado en las propiedades de los materiales que necesitan sus piezas. Esto permitiría a los equipos diseñar más libremente, permitiéndoles realizar cambios rápidamente en los productos en función de las variaciones en el coste, el suministro, los requisitos reglamentarios o las demandas de los clientes.
Por lo tanto, los beneficios del modelado y simulación de materiales multiescala se extienden más allá del valor comercial o científico puro; mejoran de manera integral la productividad en I + D. Esto permite a los científicos explorar activamente nuevas áreas de investigación de vanguardia y metodologías de materiales, que incluyen nanomateriales, biomimetismo o materiales inteligentes, entre otros.
- Detalles
- Categoría: Otras
- Visto: 7159
La tendencia continua hacia la digitalización de datos en el laboratorio produce cada vez más adeptos este año. El impulso de “no usar papeles” es una iniciativa estratégica que ofrece beneficios operativos demostrables para mejorar la productividad, reducir los tiempos de ciclo y permitir a las organizaciones aprovechar los datos experimentales y operacionales generados a lo largo el proceso de fabricación, investigación y desarrollo. Las organizaciones que buscan capturar y aprovechar estos datos de manera efectiva están explorando estas soluciones.
Datos digitalizados
Cuando los instrumentos de laboratorio empezaron a producir archivos digitales, simplemente se almacenaron en discos duro de los ordenadores locales. Con capacidades de almacenamiento y redes extraíbles, los archivos se pueden recopilar y compilar en un repositorio básico basado en carpetas. Si bien muchos lo consideran obsoleto, la realidad es que algunos laboratorios dependen del almacenamiento local de archivos para la simplicidad y la rentabilidad, al considerar los otros sistemas más modernos y automatizados demasiado complejos y costosos. Lo contrario también es cierto, muchas empresas están dispuestas a invertir en soluciones de almacenamiento de datos más avanzadas para aprovechar las capacidades específicas y preservar y reutilizar los datos en el futuro.
El auge de los sistemas informáticos de laboratorio, como los cuadernos electrónicos de laboratorio, los sistemas de gestión de información y los sistemas de ejecución, han fomentado la proliferación de información digital generada por el laboratorio.
Gestión de datos y estandarización
En la mayoría de los laboratorios, hay una gran variedad de instrumentos analíticos de diferentes proveedores, todos creando datos en varios formatos, a menudo exclusivos. El análisis y/o la combinación de datos para cualquier tipo de análisis es un proceso que requiere mucho tiempo y, a menudo implica pasos manuales y transformaciones de estos.
Al analizar los datos en un formato común o estandarizado, gracias a programas o software como los que están siendo desarrollados por algunos consorcios, puede evitarse procesos manuales y errores de transcripción. Además, los datos estandarizados son naturalmente más fáciles de combinar e interpretar junto con archivos de datos similares en el futuro.
Una vista hacia el futuro
Al igual que con muchas otras soluciones informáticas de laboratorio, la tendencia actual también es hacia la nube. En algunos casos, la totalidad del proyecto se puede alojar en la nube, desde grandes paquetes de información hasta las aplicaciones de análisis e informática del laboratorio, pero una opción bastante popular es el almacenamiento de datos local con parte del ordenador alojado como una nube. Estas opciones ponen en mano del investigador dos caminos, ambos válidos, para poder controlar el acceso a sus datos y la utilización de estos, siendo cada día más sencillo simplificar los tiempos de trabajo gracias a estas aplicaciones.
¿Aún no sabes cómo sacar el máximo beneficio de tus datos? Nosotros podemos darte la solución.
- Detalles
- Categoría: Comsol
- Visto: 7056
Ponencias y presentaciones de la conferencia COMSOL 2018.
En la Conferencia de COMSOL 2018, científicos e ingenieros de todo el mundo presentaron sus proyectos de simulación multifísica. Vea cómo los investigadores de su industria están utilizando actualmente el modelado numérico para optimizar los diseños, optimizar los procesos y mejorar los productos. Navegue por los documentos técnicos y las presentaciones utilizando el enlace inferior. Use la herramienta de búsqueda rápida para encontrar una presentación o filtre específicamente por el área de aplicación o la ubicación de la conferencia.