
BIOVIA Materials Studio
DESCRIPCIÓN
Modelado y simulación de materiales de próxima generación
BIOVIA Materials Studio es un entorno completo de modelado y simulación diseñado para permitir a los investigadores en ciencia y química de materiales predecir y comprender las relaciones de la estructura atómica y molecular de un material con sus propiedades y comportamiento. Con Materials Studio, los investigadores de muchas industrias están diseñando materiales de mejor rendimiento de todo tipo, incluidos catalizadores, polímeros, compuestos, metales, aleaciones, productos farmacéuticos, baterías y más.
Exploración de las propiedades de los materiales
Materials Studio ofrece un enfoque "in silico first", lo que permite a los investigadores optimizar el rendimiento de sus materiales en un entorno de costo relativamente bajo antes de las pruebas físicas.
-
Acelerar el proceso de innovación
Impulsar una comprensión más profunda de las interacciones que definen las propiedades de los materiales -
Reducir los costos de investigación y desarrollo
Minimice el número de experimentos físicos a través de la "selección virtual" de candidatos -
Mejorar la eficiencia de la investigación y el desarrollo
Automatice y comparta las mejores prácticas dentro de Pipeline Pilot para reducir las tareas sin valor agregado -
Fomentar las decisiones basadas en datos
Complemente la experimentación de laboratorio con una potente informática de materiales
Materials Studio incluye un entorno gráfico de usuario —Materials Studio Visualizer— en el que los investigadores pueden construir, manipular y visualizar modelos de moléculas, materiales cristalinos, superficies y estructuras mesoescalares. Materials Studio Visualizer se complementa con un extenso conjunto de métodos de solución que incluye cuántica, atomística (o clásica) mesoescalar y estadístico, que permiten a los investigadores evaluar materiales con varios tamaños de partículas y escalas temporales. También incluye herramientas para evaluar estructuras de cristales y crecimiento de cristales.
SECTORES
Polímeros y compuestos
Innovación en materiales compuestos: Perspectivas atómicas, gran impacto

Materiales como los polímeros reforzados con fibra de carbono (CFRP) proporcionan la tremenda relación resistencia-peso necesaria para su uso en aviones, satélites espaciales, turbinas eólicas, automóviles deportivos, producción submarina de petróleo y equipos deportivos. Pero su uso está limitado por los costos de fabricación, por lo que los compuestos rellenos de vidrio más baratos se utilizan en aplicaciones menos exigentes. Otro reto es la sostenibilidad: al final de su vida útil, las fibras son difíciles de extraer y reciclar. Los compuestos de caucho son otro material compuesto importante, donde los investigadores están tratando de optimizar la formulación, los rellenos y los aditivos para mejorar el desgaste de los neumáticos, la tracción y la resistencia a la rodadura.
Polímeros
- Predecir el comportamiento de polímeros puros y sus propiedades, como la temperatura de transición vítrea (Tg), el módulo de Young, el límite elástico y la deformación crítica
- Simulación de reticulación para comprender la formación de redes poliméricas y el impacto de la estructura química, los aditivos y el procesamiento en las propiedades mecánicas y la Tg
- Cálculo de energías de reacción y cinética para reacciones de polimerización y degradación
- Explore las características de los catalizadores, como la selectividad estereoquímica
- Predicción de propiedades termodinámicas con Biovia COSMOtherm
- Utilice modelos de relación cuantitativa estructura-propiedad (QSPR) para correlacionar la estructura de la unidad de repetición del polímero con propiedades a granel como Tg, relación de Poisson, conductividad térmica, índice de refracción, tensión de fractura y permeabilidad
Composites
- Resistencia cohesiva y fallo mecánico de la interfaz polímero-relleno
- Agentes de encolado de pantallas para la producción de fibras de carbono de la forma y el tamaño deseados
- Simular la reticulación de la resina alrededor de las fibras y caracterizar la estructura y la fuerza de unión
Modelado multiescala
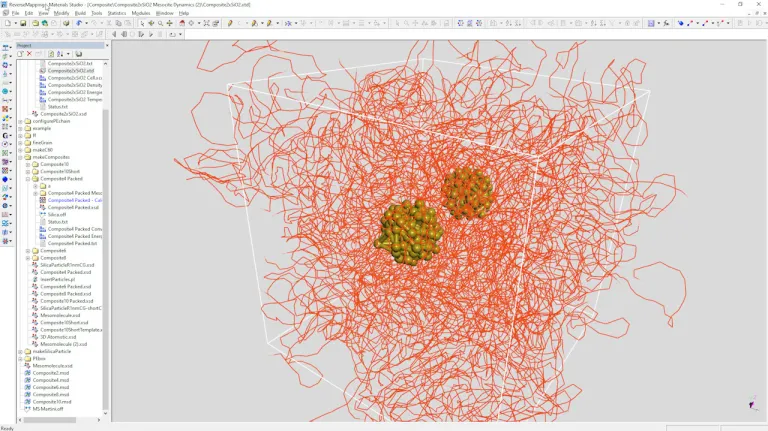
- Simule la microestructura del polímero a través de la simulación de grano grueso, como los microdominios que se forman en los copolímeros en bloque o con la adición de endurecedores termoplásticos
- Prediga las formulaciones ideales de mezclas de polímeros con métodos nativos de aprendizaje automático y flujos de trabajo de optimización multiobjetivo
- Exporte estructuras y parametrice los modelos RVE de SIMULIA Abaqus para simular materiales compuestos y piezas completas
Productos químicos y catalizadores
Acelere el desarrollo de productos con BIOVIA Materials Studio

Predicción de las propiedades de los catalizadores
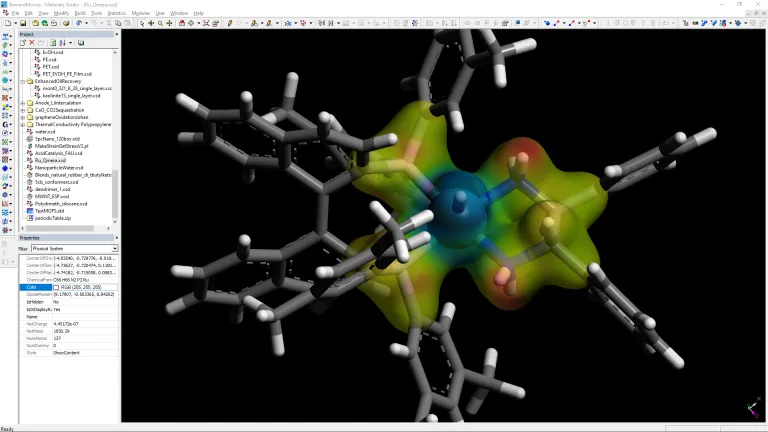
- Localice los sitios de adsorción más estables en materiales como las zeolitas
- Calcular los impactos de geometrías y/o defectos locales en el mecanismo de acción del catalizador
- Cribar nuevos catalizadores en función de sus estructuras químicas a través de métodos estadísticos y de aprendizaje automático
- Evaluar las propiedades electrónicas y químicas de los complejos metal-ligando
- Predecir la estructura y las propiedades de los catalizadores para grandes sistemas, como las nanopartículas (>500 átomos)
Modelado de reacciones

- Predecir las energías de reacción, las barreras de activación y las constantes de velocidad cinética
- Realizar simulaciones de reacciones químicas que tienen lugar en superficies cristalinas
- Predecir las concentraciones de reactivos y productos en diversas condiciones y geometrías del reactor
Especialidades químicas
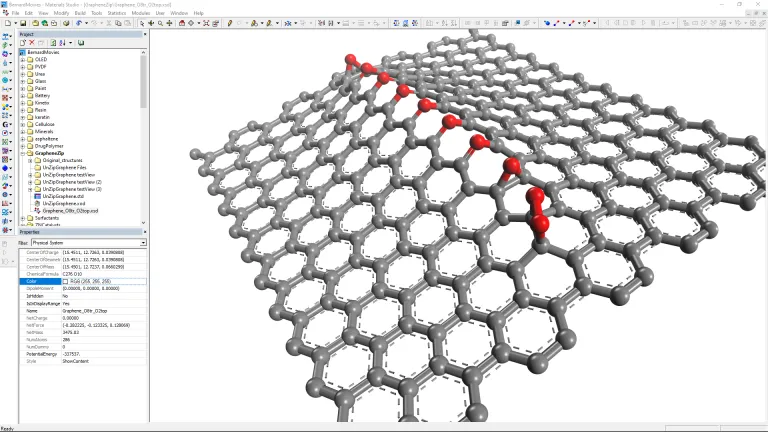
- Construir modelos predictivos que correlacionen los descriptores moleculares con las propiedades de los materiales
- Predecir las propiedades termofísicas de polímeros, tensioactivos y productos químicos a granel
- Diseñe recubrimientos protectores mediante la comprensión de los procesos químicos de la superficie, como la absorción y la corrosión a escala atómica
- Predecir las propiedades a gran escala de mezclas de productos químicos como lubricantes y mezclas de polímeros con modelos cuantitativos de relación estructura-propiedad
Productos químicos y disolventes a granel
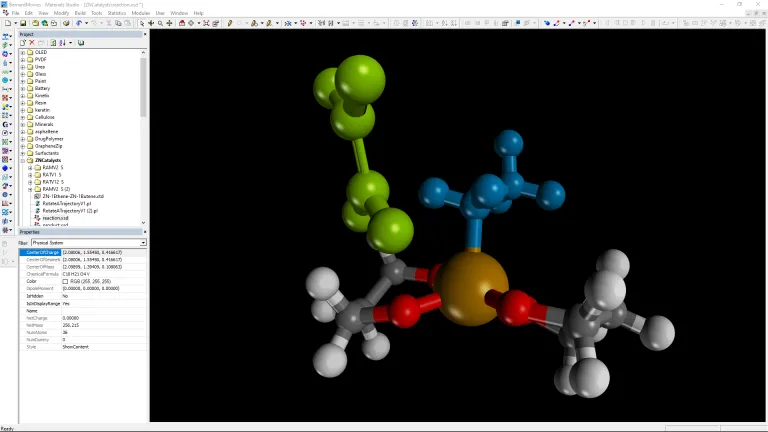
- Determinar cómo los cambios en la estructura del catalizador pueden afectar la eficiencia general de la producción de productos químicos a granel
- Comprender los mecanismos de reacción química para optimizar los procesos químicos.
- Modele las reacciones a través de una superficie de catalizador sólido para optimizar la cinética de reacción para un proceso de conversión química
- Realizar simulaciones cinéticas de reacción para predecir concentraciones de reactivos y productos en varios modelos, condiciones y geometrías de reactor y combustión
- Calcule otras propiedades termodinámicas, como la solubilidad y la presión de vapor, con BIOVIA COSMOtherm
Metales y aleaciones
Diseño de metal y aleación de silico

Aleaciones
- Proponer estructuras de celosía estables para aleaciones multicomponentes en función de su composición
- Calcular las propiedades de los materiales para las aleaciones basadas en métodos de mecánica cuántica en CASTEP, como las propiedades mecánicas y elásticas; coeficientes elásticos, módulos elásticos; propiedades térmicas, conductividad térmica, expansión térmica, capacidad calorífica; propiedades eléctricas, momento magnético
- Evaluar las interacciones moleculares entre las superficies de aleación y los recubrimientos
- Construya expansiones de clúster para aleaciones multicomponente y calcule las propiedades para cribar una gran variedad de configuraciones de aleaciones
Fabricación aditiva
- Predecir la favorabilidad energética de la formación de aleaciones desordenadas en función de la composición
- Determinar la conductividad térmica y la expansión térmica, que se pueden utilizar para comprender el comportamiento del material durante la fusión selectiva por láser y el posterior proceso de enfriamiento para evaluar la usabilidad del material para la fabricación aditiva
Productos químicos y catalizadores
Impulsando la innovación en semiconductores: de los átomos a los dispositivos

Modeling & Simulation apoya el desarrollo de estos componentes electrónicos, desde semiconductores a nanoescala hasta transistores orgánicos flexibles de película delgada y biosensores, mediante el uso de simulaciones de estructuras electrónicas como parte del diseño de dispositivos. Igual de importante es aumentar la fiabilidad de los circuitos integrados mediante el modelado de las propiedades estructurales, electrónicas, térmicas y mecánicas de los materiales.
Un conjunto completo de herramientas de modelado y simulación
BIOVIA Materials Studio proporciona un conjunto completo de herramientas de modelado y simulación para explorar las propiedades clave de los materiales que sustentan la función de los semiconductores, los transistores y la energía fotovoltaica.
Fabricación de semiconductores
- Dilucidar las características del dispositivo que definen las propiedades electrónicas
- Determinar el impacto de los defectos en el rendimiento
- Evaluación de la interfase óxido-óxido
- Evaluación y diseño de ALD, precursores de CVD
- Determinación de la densidad de estados y de la banda prohibida (con desplazamiento)
- Análisis de fotorresistencia y análisis de difusión de gases
- Transporte de electrones para generar una curva IV para ayudar al diseño de dispositivos
- Propiedades ópticas para la capacidad de detección
- Predicción de espectros de emisión de materiales de transistores para su uso en tecnologías de visualización
- Diseño de transistores de película delgada para conformar los efectos de la pulverización catódica en forma de cristal
Embalaje de semiconductores
- Determinar la heterogeneidad mesoscópica en el proceso de curado de resinas para evaluar su expansión térmica durante las etapas de curado
- Calcule la adhesión de las resinas epoxi con las superficies metálicas para abordar los problemas de estabilidad y dependencia de la humedad
- Propiedades estructurales y electrónicas de las uniones soldadas
Baterías y pilas de combustible de hidrógeno
Diseño avanzado de materiales para aumentar el rendimiento de la batería

Acelere el diseño de fuentes de energía sostenibles
BIOVIA Materials Studio apoya la caracterización y el desarrollo de materiales existentes y novedosos para baterías y pilas de combustible. Junto con la cartera más amplia de Dassault Systèmes, los científicos de materiales pueden acelerar el diseño de fuentes de energía y almacenamiento sostenibles, seguros y fiables.
Ánodo
- Predecir el transporte de iones de litio en el ánodo y cómo se organizan los iones de litio dentro del ánodo durante la carga y descarga
- Determinar los procesos que conducen a la exfoliación del grafeno y a la degradación general del ánodo
- Explorar los factores que controlan el crecimiento y la estabilización de la interfase electrolítica sólida
- Determinar los mecanismos a escala atómica para la degradación en el rendimiento de la batería, como la formación de dendritas metálicas
Cátodo
- Calcule el voltaje de celda abierta (OCV) para varios materiales de cátodo para estimar las curvas de carga y descarga
- Evaluar los cambios en la estructura del cátodo durante el ciclo para determinar su impacto en la capacidad total del cátodo
- Explore y optimice las interacciones entre la superficie del cátodo y varios recubrimientos
Electrólito
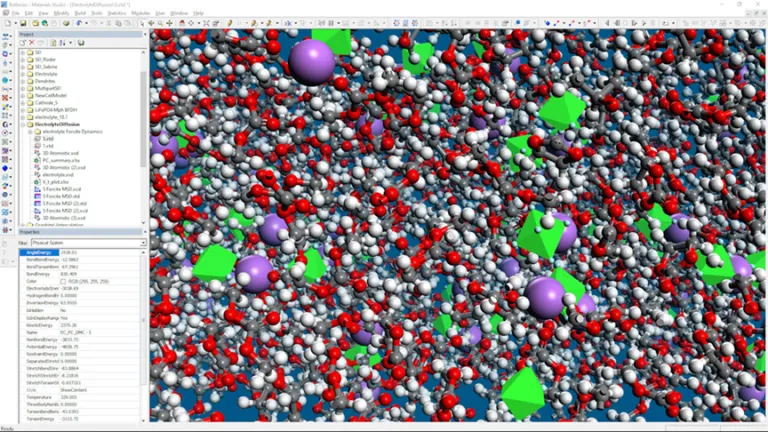
- Calcular la difusión de iones de litio a través de electrolitos puros o mezclas
- Investigar cómo se rompen las moléculas de electrolito y se incorporan a la interfaz de electrolito sólido durante el funcionamiento
- Determinar las propiedades de los aditivos electrolíticos para evaluar cómo influyen en el rendimiento
- Predecir la viscosidad de las formulaciones de electrolitos
- Materials Studio también puede combinarse con COSMOtherm para predecir la seguridad de los electrolitos en función de propiedades como la presión de vapor y el punto de inflamación
Pilas de combustible de hidrógeno y electrolizadores
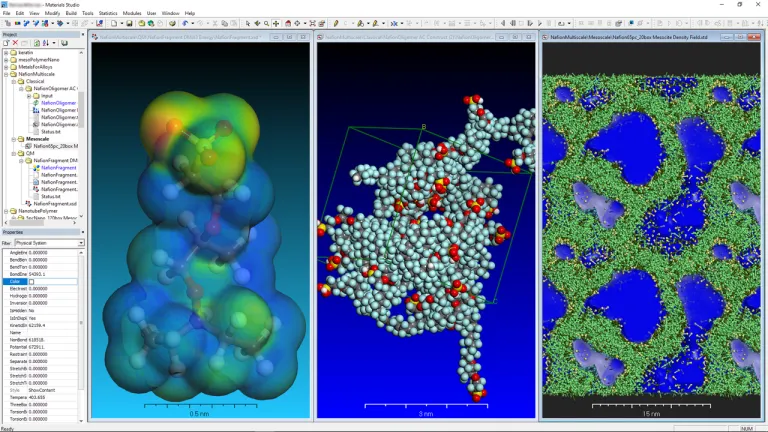
- Optimice la durabilidad y el rendimiento de las membranas poliméricas mediante predicciones de la morfología y el impacto en la difusión de protones y el transporte de agua.
- Comprender los mecanismos de degradación de la membrana
- Reduzca los costos mediante la detección de materiales de electrodos alternativos
- Suministre datos e información sobre materiales a elementos finitos (SIMULIA Abaqus) y modelos de sistemas (CATIA Dymola) de conjuntos de pilas de combustible y electrolizadores
Electrónica
Modelado de Materiales para la Innovación Electrónica

BIOVIA Materials Studio apoya la caracterización y el desarrollo de materiales existentes y novedosos para exhibición, incluidos OLED, semiconductores orgánicos y más.
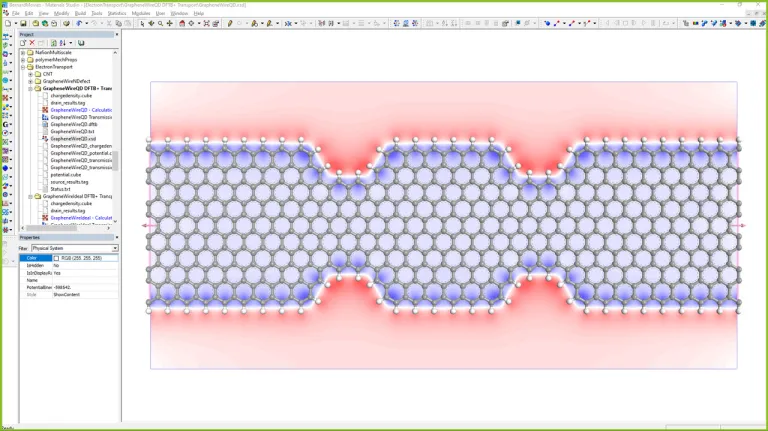
Las peroskovitas como reemplazo de las células fotovoltaicas tradicionales basadas en silicio prometen una conversión más eficiente de la energía luminosa en electricidad y opciones de fabricación significativamente más flexibles. Sin embargo, sigue habiendo desafíos para extender la vida útil de estos materiales, que son propensos a la degradación, y para reemplazar los componentes tóxicos. Los OLED, como fuentes de luz y como tecnologías de visualización, necesitan desarrollos en múltiples áreas para aumentar la eficiencia, la vida útil y el rendimiento y reducir los costos de fabricación. El desarrollo de materiales emisores azules que permitan mejorar la estabilidad y la eficiencia, y los métodos para extraer de manera eficiente la luz generada son dos desafíos clave.
OLED
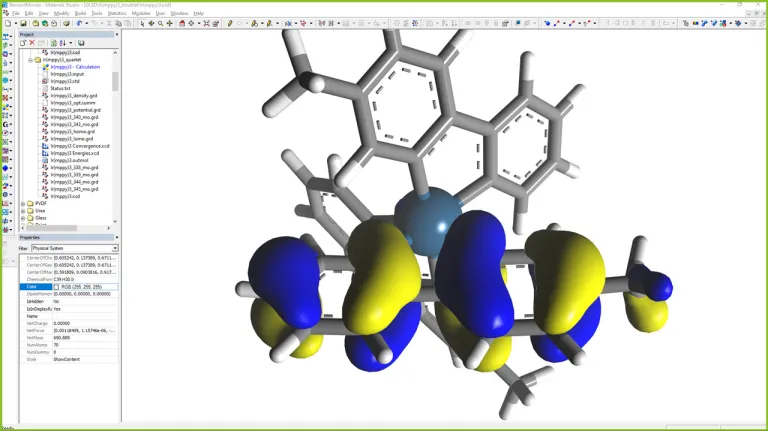
- Predicción de la luminiscencia de las fotos
- Predicción de fluorescencia y fosforescencia
- Cálculo del transporte de electrones utilizando los métodos de la función de Green para monitorizar la curva IV tanto para estructuras periódicas como no periódicas
- Cálculo de la función de transmisión en la interfaz
- Transporte de carga y movilidad de portadores en fotodetectores orgánicos
- Proceso de generación y separación de carga en heterounión a granel
- Cálculo del desplazamiento de banda y de la movilidad para sensores metálicos impregnados de metal
- Polarizabilidad e hiperpolarizabilidad de los compuestos orgánicos adsorbidos sobre superficies activas
- Confirmación de la estructura de OLED o semiconductores orgánicos mediante simulación de RMN de estado sólido
- Capacidad para diseñar el modelo intercalado completo del electrodo y la región del dispositivo orgánico
- Flujos de trabajo automatizados de alto rendimiento basados en Python para calcular el estado fundamental y las propiedades del estado excitado para generar automáticamente una base de datos
Fotovoltaica y Perovskitas
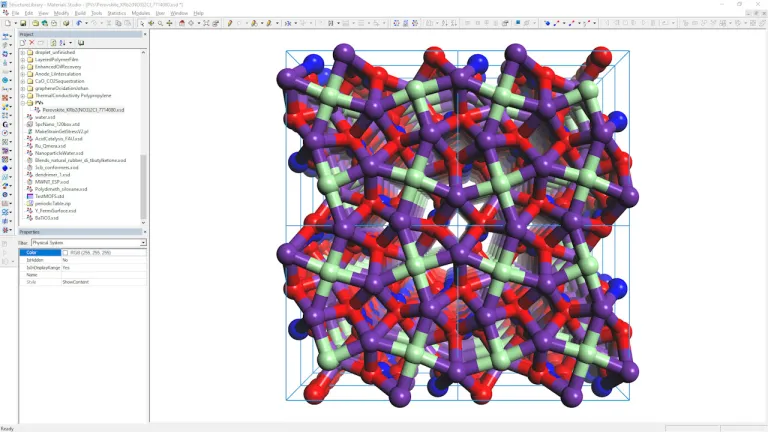
- Fijación de la capa activa con vidrio
- Simular posibles mecanismos de grabado
- Comprender la deposición, la impresión y la adhesión a sustratos
- Predicción de espectros ópticos mediante cálculos de primeros principios
- Predicción de la solubilidad de los materiales en el procesamiento de disolventes con COSMOtherm
- Simular el curado de películas poliméricas para encapsulación
- Predicción de la migración de agujeros y una recombinación utilizando ab initio MD
- Influencia del orden molecular en la movilidad de los portadores de carga
Bienes de consumo envasados
Diseñe mejores productos de consumo con simulación

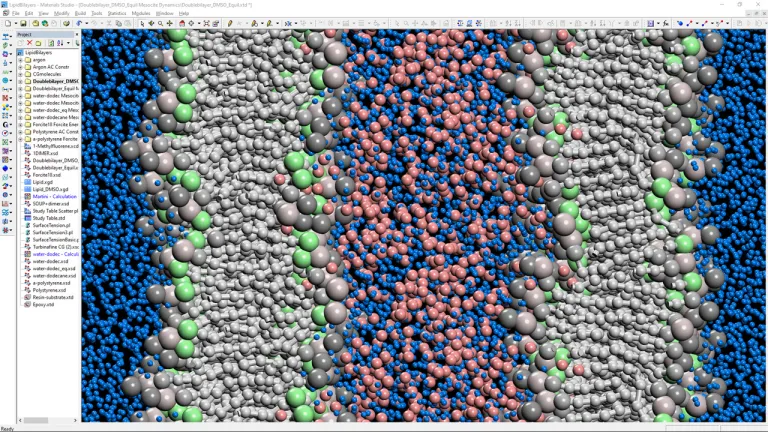
Acelere el desarrollo de productos
BIOVIA Materials Studio proporciona un conjunto completo de soluciones para el diseño y desarrollo de productos de bienes de consumo mediante la comprensión de las interacciones atómicas que sustentan el comportamiento del producto. El uso de modelos y simulaciones para guiar los proyectos de investigación y desarrollo acelera significativamente el desarrollo de productos, desde la comprensión de las propiedades de la espuma de un nuevo champú hasta la cuantificación de la permeabilidad de los tintes en un producto para el cuidado de la belleza. Las empresas pueden centrar sus experimentos de laboratorio en candidatos más prometedores que tengan más probabilidades de satisfacer las expectativas de los consumidores.
Alimentos y bebidas

- Explore los impactos de las condiciones ambientales en la frescura y la apariencia de los alimentos, como la floración del chocolate
- Evaluar los impactos de la sustitución de grasas y aceites de origen vegetal o "verdes" por modelos cuantitativos de relación estructura-propiedad (QSPR)
- Prediga las propiedades de las formulaciones de los productos y optimícelas para los mercados finales con flujos de trabajo nativos de aprendizaje automático
- Genere modelos de farmacoforos para predecir nuevos olores y saborizantes con la herramienta CATALYST en BIOVIA Discovery Studio
Cuidado personal

- Prediga la permeabilidad cutánea de los ingredientes activos y aditivos con BIOVIA COSMOplex
- Calcule la solubilidad con BIOVIA COSMOtherm
- Determinar los espectros de emisión y absorción de moléculas de pigmento
- Identifique las características moleculares que imparten propiedades deseables para el consumidor, como la formación de espuma y la estabilidad, con modelos QSPR
Productos para el hogar

- Predecir la formación de micelas para evaluar la actividad y el comportamiento del detergente
- Determinar los impactos de la sustitución de ingredientes por alternativas "verdes"
- Calcule propiedades termodinámicas como la presión de vapor de disolvente con BIOVIA COSMOtherm para determinar su uso en aerosoles y pulverizaciones
Embalaje

- Predecir las propiedades de polímeros y mezclas para optimizarlos para el envasado y transporte de alimentos
- Determinar los productos de degradación de los envases debido al medio ambiente
- Calcule la resistencia de los adhesivos y comprenda el mecanismo de unión
Desarrollo farmacéutico
Modelado de fármacos in silico

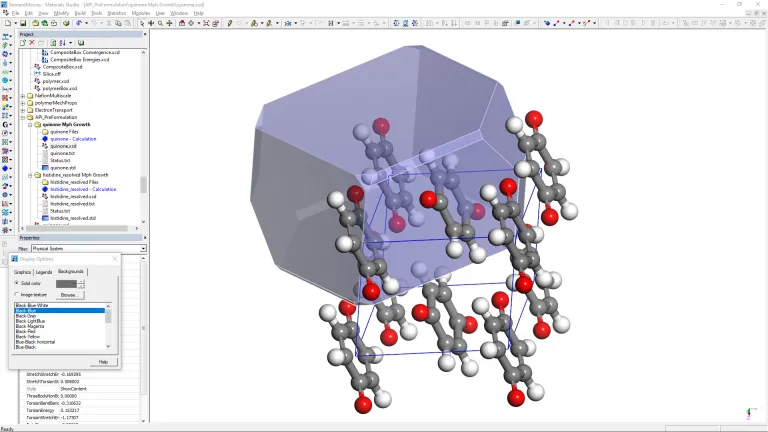
Una suite completa
BIOVIA Materials Studio proporciona un conjunto completo de métodos para modelar el comportamiento de los compuestos farmacológicos in silico, lo que puede ayudar a guiar los esfuerzos de desarrollo, minimizando el riesgo y respaldando la captura de propiedad intelectual.
Cristalización
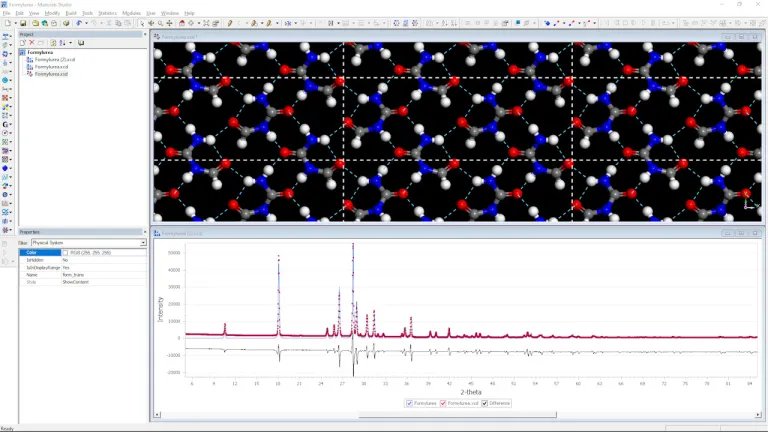
- Determinar las estructuras cristalinas de los compuestos farmacológicos a través de espectros de difracción de polvo simulados y refinarlos en función de los resultados experimentales
- Explorar posibles polimorfos de un compuesto directamente a partir de su estructura molecular
- Predecir la morfología de un material cristalino a partir de su estructura cristalina y explorar los efectos de los disolventes y las impurezas en el crecimiento de las facetas cristalinas
Estabilidad
- Determinar el potencial de los compuestos candidatos para la autooxidación o las reacciones cruzadas con diversos excipientes
- Predecir la temperatura de transición vítrea y otras propiedades para explorar la estabilidad de compuestos candidatos en dispersión de sólidos amorfos
- Determinar las propiedades mecánicas de los compuestos candidatos para su fabricación (molienda, mezcla, etc.)
Solubilidad
- Lleve a cabo cálculos de alto rendimiento de una variedad de coeficientes de dispersión, como logD y AlogP
- Cálculo de los parámetros de solubilidad y miscibilidad de los compuestos mediante simulación
- Predecir la solubilidad de los compuestos en varios disolventes y cocristales de cribado
- Determinar la "carga" de compuestos en micelas y otros sistemas portadores
CARACTERÍSTICAS
HERRAMIENTAS CUÁNTICAS
Materials Studio proporciona una gama de solucionadores basados en la teoría funcional de la densidad y métodos semiempíricos para predecir propiedades de materiales moleculares y de estado sólido a partir de estructuras electrónicas. También incluye solucionadores especializados para utilizar energías y propiedades derivadas de la mecánica cuántica en reacciones químicas.
| PRODUCTO | DESCRIPCIÓN |
| Materials Studio CANTERA | Cantera [www.cantera.org] es un solucionador de ecuaciones de tasas químicas. Materials Studio Cantera proporciona un entorno para configurar la entrada termodinámica y para ejecutar estos cálculos. "Cantera Reaction Editor" permite a los usuarios introducir nuevas especies y reacciones, opcionalmente con velocidades de reacción determinadas a partir de Materials Studio DMol3, en esquemas de reacción complejos con datos termodinámicos existentes determinados experimentalmente. |
| Materials Studio CASTEP | Simula las propiedades de sólidos, interfaces y superficies para una amplia gama de materiales, incluidos cerámicas, semiconductores y metales, utilizando un método funcional de densidad de onda plana. |
| Materials Studio DMol3 | Se utiliza para modelar la estructura electrónica y las propiedades de moléculas orgánicas e inorgánicas, cristales moleculares, sólidos covalentes, sólidos metálicos y superficies infinitas utilizando DFT. |
| Materials Studio DFTB+ | Módulo semiempírico para simular propiedades electrónicas de materiales. Utiliza un enfoque de vinculación estricta basado en la teoría funcional de la densidad para permitir la precisión de la mecánica cuántica en sistemas de mayor tamaño. |
| Materials Studio FlexTS | Herramienta sólida para identificar la ruta de energía mínima entre reactivos y productos en reacciones químicas (ubicación de estados de transición). FlexTS requiere DMol3 o DFTB+ para suministrar la energía del sistema de cada configuración |
| Materials Studio KINETIX | Programa de propósito general para simular los procesos químicos y físicos competitivos de adsorción, desorción y difusión que tienen lugar en las superficies. Esto proporciona información única, como el papel de la difusión de especies en la actividad y el envenenamiento de los catalizadores, y la cobertura superficial de especies hasta en microescalas. |
| Materials Studio NMR CASTEP |
Predice cambios químicos de RMN y tensores de gradiente de campo eléctrico a partir de los primeros principios. El método se puede aplicar para calcular los cambios de RMN de moléculas y sólidos para una amplia gama de materiales, incluidos cerámicos y semiconductores. |
| Materials Studio ONETEP | Código DFT de escala lineal que permite cálculos precisos de primeros principios en sistemas de hasta miles de átomos. |
| Materials Studio QMERA | Emplea el método QM/MM que combina la precisión de un cuanto con la velocidad de un cálculo de campo de fuerza. Este enfoque permite realizar cálculos precisos en sistemas muy grandes con un esfuerzo sustancialmente menor. |
| Materials Studio VAMP | Predece rápidamente muchas propiedades físicas y químicas de sistemas moleculares orgánicos e inorgánicos utilizando un método de orbitales moleculares semiempírico. Materials Studio VAMP es un enfoque intermedio ideal entre los métodos de campo de fuerza y primeros principios. |
HERRAMIENTAS DE SIMULACIÓN CLÁSICAS
Materials Studio ofrece una amplia gama de métodos basados en interacciones clásicas entre átomos y moléculas. Éstas incluyen
Dinámica molecular, dinámica de celosía y varios métodos basados en Monte Carlo, así como el suministro de campos de fuerza.
| PRODUCTO | DESCRIPCIÓN |
| Materials Studio Adsorption Locator |
Encuentra sitios de adsorción de baja energía para moléculas en sustratos periódicos y no periódicos. |
| Materials Studio Amorphous Cell |
Conjunto de herramientas computacionales que le permiten construir modelos representativos de sistemas amorfos complejos y predecir propiedades clave. |
| Materials Studio Blends | Predice diagramas de fases y parámetros de interacción para mezclas líquido-líquido, polímero-polímero y aditivos poliméricos, equilibrios de fases y tecnología de separaciones. |
| Materials Studio Conformers |
Proporciona algoritmos de búsqueda conformacional y herramientas de análisis para caracterizar la conformación y flexibilidad molecular. |
| Materials Studio COMPASS |
Predicción precisa de propiedades estructurales, conformacionales, vibratorias y termofísicas para una amplia gama de moléculas aisladas y en fases condensadas, y bajo una amplia gama de condiciones de temperatura y presión. Esto incluye acceso a los últimos parámetros de COMPASS III (https://doi.org/10.1080/08927022.2020.1808215), altamente validados y que cubren la más amplia gama de materiales. |
| Materials Studio Forcite Plus |
Ofrece métodos de dinámica y mecánica molecular para moléculas y sistemas periódicos. La herramienta incluye una amplia gama de funciones de análisis para predecir propiedades mecánicas, difusividad, estructura local, variaciones de densidad, densidad de energía cohesiva, autocorrelación dipolar funcional y más. Los campos de fuerza admitidos son Materials Studio COMPASS, CVFF, PCFF, Dreiding y Universal. Forcite Plus también admite la ejecución en GPU para un rendimiento acelerado. |
| Materials Studio GULP | Método de optimización, cálculo de propiedades y dinámica de materiales. Incluye una amplia gama de campos de fuerza para metales, óxidos, minerales semiconductores, así como campos de fuerza de mecánica molecular para sistemas covalentes. También se proporcionan herramientas de ajuste Forcefield para desarrollar parámetros para materiales personalizados. |
| Materials Studio Sorption | Proporciona un medio para predecir propiedades fundamentales necesarias para investigar fenómenos de adsorción y separaciones, como las isotermas de sorción y las constantes de Henry. |
HERRAMIENTAS DE SIMULACIÓN EN MESOESCALA
Los métodos de mesoescala en Materials Studio se basan en dos enfoques de grano grueso (1) donde los grupos de átomos se reemplazan por cuentas o (2) donde las regiones de materiales se representan mediante campos de densidad. Utilizando estos enfoques es posible ampliar las escalas de longitud y tiempo accesibles en varios órdenes de magnitud con respecto a las simulaciones clásicas.
| PRODUCTO | DESCRIPCIÓN |
| Materials Studio MesoDyn | Método funcional de densidad clásico para estudiar el comportamiento a largo plazo y escala de tiempo de sistemas de fluidos complejos, en particular la separación de fases y la estructura de sistemas poliméricos complejos. |
| Materials Studio Mesocite | Módulo de simulación de grano grueso para el estudio de materiales en escalas de longitud que van desde nanómetros a micrómetros y escalas de tiempo de nanosegundos a microsegundos. Materials Studio Mesocite puede proporcionar propiedades estructurales y dinámicas de fluidos en equilibrio, bajo corte o en geometrías confinadas. |
| Materials Studio PhaseField |
Módulo que proporciona predicciones de la microestructura en materiales duros, como la estructura del grano en aleaciones metálicas complejas, mediante la simulación de la solidificación y el crecimiento del grano. Este módulo se ejecuta utilizando protocolos proporcionados por Pipeline Pilot Materials Studio Collection |
HERRAMIENTAS ESTADÍSTICAS
Las herramientas estadísticas son ideales para detectar compuestos rápidamente al relacionar los rasgos moleculares directamente con las cantidades observadas experimentalmente.
| PRODUCTO | DESCRIPCIÓN |
| Materials Studio QSAR | La integración de QSAR (Relaciones cuantitativas estructura-actividad) en Materials Studio brinda acceso a una amplia gama de descriptores y capacidades de análisis avanzadas para ayudar a generar relaciones estructura-actividad de alta calidad. QSAR incluye una amplia gama de descriptores, incluidos descriptores topológicos y electrotopológicos. Además, los descriptores Jurs permiten examinar la distribución de carga en las superficies del disolvente; Los descriptores VAMP amplían aún más la gama de descriptores 3D a aquellos que incluyen interacciones electrónicas; y GFA aplica una genética sofisticada para calcular relaciones cuantitativas estructura-actividad. |
| Materials Studio QSAR Plus |
QSAR Plus agrega el poder de los descriptores DMol3 para calcular índices de reactividad y energías precisas a QSAR. También se incluyen redes neuronales para construir modelos no lineales y modelos que sean más resistentes a conjuntos de datos ruidosos que otros métodos de construcción de modelos. También se puede utilizar con conjuntos de datos a los que les faltan algunos valores y se puede utiliza para construir modelos ponderados para predecir múltiples propiedades físicas. |
| Materials Studio Synthia | Synthia calcula las propiedades de homo y copolímeros utilizando relaciones cuantitativas estructura-propiedad (QSPR) avanzadas. Permite a los investigadores seleccionar rápidamente polímeros candidatos para una amplia gama de propiedades. |
HERRAMIENTAS DE ANÁLISIS Y CRISTALIZACIÓN
Se emplean herramientas analíticas y de cristalización para investigar, predecir y modificar la estructura cristalina y el crecimiento de los cristales.
| PRODUCTO | DESCRIPCIÓN |
| Materials Studio Morphology |
La morfología le permite predecir la morfología del cristal a partir de la estructura atómica de un cristal. La morfología permite la predicción de la forma del cristal, el análisis de la estabilidad de la superficie del cristal, el desarrollo de aditivos personalizados y el control de los efectos de los disolventes y las impurezas. |
| Materials Studio Polymorph Predictor |
Polymorph Predictor ha sido desarrollado para su uso con moléculas iónicas o no iónicas bastante rígidas compuestas principalmente de carbono, nitrógeno, oxígeno e hidrógeno. El enfoque se basa en la generación de posibles disposiciones de empaquetamiento en todos los grupos espaciales razonables para buscar los mínimos bajos en la energía de la red. |
| Materials Studio Motif | Motif analiza información de conectividad en cristales moleculares, proporcionando un método de análisis cualitativo y cuantitativo de topologías de enlaces de hidrógeno. Combinado con las capacidades predictivas de Polymorph, Motif permite la categorización y puntuación estadística de las estructuras propuestas. Interactúa con la base de datos estructural de Cambridge aprovechando la funcionalidad Mercury del Centro de datos cristalográficos de Cambridge. |
| Materials Studio Reflex | Reflex simula patrones de difracción de polvo de rayos X, neutrones y electrones basados en modelos de materiales cristalinos. Reflex Plus ofrece un paquete completo para la determinación de estructuras cristalinas a partir de datos de difracción de polvo de calidad media a alta. |
| Materials Studio Reflex QPA |
Reflex QPA amplía la funcionalidad de Reflex para el análisis de fases cuantitativo, permitiendo la determinación de la proporción relativa de diferentes fases, incluidos sistemas tanto inorgánicos como orgánicos, en una mezcla basada en datos de difracción de polvo. |
| Materials Studio X-Cell | X-Cell es un algoritmo de indexación eficiente para datos de difracción de polvo de calidad media a alta. X-Cell utiliza un procedimiento de dicotomía específico de extinción para realizar una búsqueda exhaustiva del espacio de parámetros para establecer una lista completa de todas las posibles soluciones de celda unitaria. |
 BIOVIA Discovery Studio
BIOVIA Discovery Studio