- Detalles
- Categoría: Comsol
- Visto: 7067
La versión de COMSOL® 5.3a update 2 contiene mejoras de rendimiento y estabilidad de COMSOL Multiphysics®, COMSOL Server™, y COMSOL Client. La actualización se aplica sobre COMSOL® versión 5.3a (Build: 180) y versión 5.3a update 1 (Build: 201). La actualización puede ser aplicada directamente sobre cualquier instalación de la versión 5.3a.
Si dispone de una versión anterior a la versión 5.3a y una licencia válida que esté en suscripción, entonces realice una instalación completa de la versión 5.3a desde la página de descarga del producto, que incluirá todas las actualizaciones.
Cómo aplicar la actualización del software
Antes de proceder con su instalación, asegúrese de que ha cerrado y salido de los procesos de COMSOL® o cualquier software que esté corriendo que se corresponda con un producto LiveLink™ que desee actualizar (Excel®, MATLAB®, o un programa CAD).
Actualización de COMSOL Multiphysics®
La manera más fácil de instalar la actualización para COMSOL Multiphysics® es arrancar el programa y seleccionar Buscar actualizaciones del producto. Si está utilizando el sistema operativo Windows®, esta opción está localizada en el menu File, bajo Ayuda. Si utilize el sistema operative Linux® o macOS, está localizada en el menú Ayuda.
Para verificar que la instalación de la actualización fue correcta, primero arranque el programa COMSOL Multiphysics® y seleccione la opción del menú Sobre COMSOL Multiphysics. Si utiliza el sistema operativo Windows®, la encontrará en el menú File bajo Ayuda. En el caso de que utilice el sistema operativo Linux® o macOS, estará localizada bajo el menú Ayuda. La versión listada debería ser COMSOL Multiphysics® 5.3a (Build: 229).
Actualización de COMSOL Server™
Para actualizar COMSOL Server™ si está utilizando el sistema operativo Windows®, corra el program Update de la carpeta COMSOL Launchers en el menú Start.
Nótese que el proceso de servicio de COMSOL Server™ tiene que ser parado antes de aplicar la actualización.
Para comprobar que la instalación se realize correctamente, primero asegúrese de que COMSOL Server™ esté corriendo, entonces navegue a COMSOL Server™ con un navegador web y siga el enlace Sobre COMSOL Server™ en la esquina inferior derecha. La nueva versión debería ser COMSOL Server™ 5.3a (Build: 229).
Actualización de COMSOL Client para su uso con COMSOL Server™
Para actualizar COMSOL Client para el sistema operative Windows®, descárguese la nueva versión desde Client Download e instale. La versión anterior de 5.3a se sobrescribirá. Su instalación de COMSOL Client deberá de tener el mismo número de versión que la instalación de COMSOL Server™ a la que se conecte.
COMSOL Multiphysics®
- Solucionado un problema al resaltar los resultados de una búsqueda en la ventana de Documentation.
- Corregido un problema que imposibilitaba copiar y pegar texto desde la ventana de Ayuda en el entorno COMSOL Desktop®.
- Añadido el nodo Malla móvil, disponible como un subnodo de Componente > Definiciones, en el árbol del modelo al seleccionar Modificar la configuración del modelo para un paso de estudio. Las opciones disponibles son: Solve for y Disable in solvers.
- Gestión mejorada de las condiciones de contorno Velocidad de malla normal prescrita en dominios deformantes. El cambio hace que los cambios de volumen de un dominio sean consistentes con la velocidad normal prescrita.
- Robustez aumentada para las funcionalidades de Dominio deformante. El método por defecto de suavizado de malla se ha cambiado de Laplace a Yeoh. El método Yeoh es más robusto con un pequeño coste computacional adicional.
- Los nodos de Malla móvil, añadidos bajo Definiciones > Malla móvil, ahora incluyen una sección Anular que contiene información sobre cómo esos nodos se anulan entre sí. También es posible mover esos nodos y copiar y pegarlos.
- Añadida la capacidad para las interfaces Matemáticas basadas en PDE de ser activas en dominios que sean parte de una funcionalidad de Capa absorbente.
- Corregido un problema con las selecciones de Geometría de forma que las que son dependientes de un parámetro mantienen la misma dependencia cuando se utilizan bajo Resultados.
- Arreglado un problema con el uso de nodos Función Cambiar que accedían a recursos de archivos.
- Corregidas las entradas de modelo definidas por el usuario de forma que ahora muestran todas las componentes en lugar de solo las componentes en el plano.
- Arreglado un problema con parámetros de búsqueda en la ventana de la Biblioteca de aplicaciones.
- Añadido el Soporte para parámetros de búsqueda basado en los resultados en la ventana de Biblioteca de aplicaciones. Ahora se puede buscar utilizando cadenzas de caracteres como @dataset, @numerical, @export, y @report.
- Ahora, al instalar el software en japonés, se encuentran disponibles versiones en idioma japonés de los documentos PDF del Software License Agreement e Introduction to COMSOL Multiphysics.
COMSOL Server™
- Mejorada la alineación de miniaturas personalizadas en la página de la Librería de aplicaciones de COMSOL Server.
Acoustics Module
- Corregido un problema con la condición de Pared de forma que la rugosidad de Rayleigh puede ser utilizada en combinación con un coeficiente de absorción definido por el usuario.
- Arreglado un problema con la definición de la variable de postprocesado módulo de volumen de fluido en la interfaz de Ondas poroelásticas.
CAD Import Module
- Solucionado un problema donde el registro automático de la función de archivo de licencia de CADREADER se retrasó en algunos entornos. La funcionalidad de archivo de licencia de CADREADER habilita la capacidad de importar archivos de datos CAD en los productos de interfaz CAD.
CFD Module
- Corregido un problema con el orden de la función de forma para ciertas variables Flujo de burbujas cuando se utilizaban en combinación con elementos P2+P2 y P3+P3.
- Corregidos unos cuantos problemas que podían ocurrir al utilizar los modelos de turbulencia específicos de fase y la interfaz Euler-Euler.
- Solucionado un problema con la condición Ventilador interno para el acoplamiento multifísico Flujo reactivo.
- Solucionado un problema con las condiciones de Ventilador interno y Tamiz para modelos de flujo no isotérmico turbulento.
Chemical Reaction Engineering Module
- Corregida la constante de tasa de reacción para reacciones de superficie importadas desde el software CHEMKIN®.
Electrochemistry Module
- Corregido un error de signo en la desviación de potencial para modelos que utilizan un nodo de Contorno de membrana de intercambio iónico.
Fatigue Module
- Corregido un problema donde los gráficos de Matriz de historgrama no se generaban para una interfaz de Daño acumulativo.
Heat Transfer Module
- Solucionado un problema con la formulación de estabilización para modelos donde una funcionalidad de Translational Motion se combinaba con un sistema de coordenadas definido por el usuario.
- Corregida la dependencia de temperatura para materiales de capas delgadas generales.
- Añadido Soporte para múltiples instancias de la funcionalidad Propiedades de medio en la interfaz física Radiative Beam in Absorbing Media.
LiveLink™ for MATLAB®
- Solucionado un problema con mphsave en sistemas operatives Windows® que impedía guardar un archivo en un disco de red no mapeado.
- Solucionado un problema con mphplot que impedía que versions más antiguas del software MATLAB® funcionara con el programa COMSOL Multiphysics® versión 5.3a.
LiveLink™ for SOLIDWORKS®
- Mejorado el rendimiento al abrir un archivo que contenga selecciones con un gran número de entidades en la selección.
Nonlinear Structural Mechanics Module
- Corregido un error en el modelo de material hiperelástico de Storakers.
Optimization Module
- Solucionado un problema que imposibilitaba resolver modelos de optimización que tuvieran pasos de estudio no basados en la física.
Ray Optics Module
- Corregidas algunas transformación de los parámetros de Stokes y fase al trazar rayos polarizados a través de medios con un índice gradual.
Semiconductor Module
- Añadido Soporte para incluir una funcionalidad de Capa perfectamente adaptada en un modelo que contenga la interfaz de Ecuación de Schrödinger con únicamente una licencia del módulo Semiconductor Module.
Structural Mechanics Module
- Mejora en la formulación de la interfaz Membrana para casos donde se seleccionó una descomposición de deformación aditiva en un estudio geométriamente no lineal. Ahora, en su lugar, se utiliza una formulación de deformación no lineal en lugar de la formulación de deformación lineal anterior.
- Corregido un error de "Variable indefinida" en la interfaz Thin Film Damping.
- Añadido soporte para deformación plana generalizada al combinar materiales piezoeléctricos y elásticos lineales.
- Corregidos algunos problemas que podian ocurrir al utilizar fricción en una funcionalidad de Bolt Thread Contact.
CHEMKIN es una marca registrada de Reaction Design Corp. en los Estados Unidos y otros países. MATLAB es una marca registrada de The MathWorks, Inc. Microsoft y Windows son marcas registradas o comerciales de Microsoft Corporation en los Estados Unidos y/o otros países. SOLIDWORKS es una marca registrada de Dassault Systèmes SolidWorks Corp.
- Detalles
- Categoría: ChemOffice
- Visto: 56346

Varios cientos de estudiantes toman la decisión de estudiar química a nivel universitario cada año. La mayoría de estos entran en este mundo con un vacío enorme a nivel orgánico que puede ser suplido por el uso de grandes herramientas de tratamiento de información química, como es ChemDraw. Los estudios previos a los universitarios podrían verse enormemente reforzados con esta interfaz, facilitando de esta forma el posterior entendimiento de la química universitaria. A continuación, se tratarán los aspectos básicos que pueden enseñarse en estudios previos universitarios.
Estructura molecular, conformación y configuración estructural
Uno de los primeros pasos en el entendimiento de la formulación química reside en saber distinguir conformación de configuración estructural, siendo necesario en el primer caso distintas orientaciones de los grupos funcionales en el espacio, pero sin necesidad de romper enlaces simples y, en el segundo caso, necesaria la ruptura de enlaces para la formación de nuevas moléculas.
Con ChemDraw pueden plantearse los distintos tipos de isómeros configuracionales para distintos tipos de moléculas. Para ello simplemente deben utilizarse los comandos de dibujo situados en la columna izquierda del panel principal de ChemDraw, bien por dibujo tradicional o mediante uso de Hotkeys.

Podemos realizar además simulaciones para estructuras sencillas (y complejas) de isómeros conformacionales. De esta forma se muestra a continuación dos ejemplos de isómeros conformacionales.
 Proyección de Newman del butano (b) Confórmeros del cis-1,3-dimetilciclohexano")
Como podemos observar, en el caso de la molécula de butano, los metilos se encuentran en la posición más alejada (180˚) para el confórmero más estable de tal forma que la interacción espacial entre ambos grupos funcionales es mínima. En el caso de la molécula de 1,3-dimetilhexano la conformación más estable sería la expuesta a la izquierda de las flechas de equilibrio, donde los metilos se disponen de forma “cis”, más alejado el uno del otro.
Dentro de los isómeros conformacionales encontramos la estereoquímica, se produce cuando el carbono que se estudia está unido a cuatro sustituyentes diferentes de tal forma que se conoce como “carbono quiral”. Los compuestos que dispongan de carbonos quirales en su estructura pueden tener actividad óptica. Estos isómeros ópticos, en pareja, son imagen especular el uno del otro, estas dos estructuras serán enantiómero la una de la otra.

Como podemos ver, ChemDraw nos ofrece la posibilidad de obtener la dirección en la que dicho carbono desviará el plano de la luz polarizada, para lo cual deberemos seleccionar la molécula y con el botón derecho utilizar la opción “Atom – Show Stereochemistry”.
Otro aspecto muy útil a la hora de trabajar con ChemDraw a nivel docente reside en la opción “nombre-estructura” que permitirá a los alumnos identificar estructuras químicas a partir de su nombre.

Además de ahorrar tiempo con este comando, puede utilizarse la opción “estructura-nombre” que nos permite responder al momento a preguntas del tipo “¿cómo nombrarías este compuesto?”.

Esta función es lo suficientemente sofisticada como para nombrar con precisión moléculas con centros proquirales.

Reacciones orgánicas, laboratorio y espectroscopía
Además de poder analizar estructuras a nivel molecular, ChemDraw es útil para plantear procesos de síntesis orgánica (así como inorgánica), procedimientos de laboratorio (gracias a sus múltiples plantillas de material de vidrio) y espectroscopía de RMN.

La biblioteca de imágenes prediseñadas hace que sea sencillo ilustrar montajes de laboratorio. Un ejemplo de esto sería una destilación de vapor simple.

Antes de pasar a niveles de educación universitarios los alumnos deberían saber interpretar espectros de resonancia magnética nuclear, tanto de protón como de carbono. Para una gran variedad de estructuras ChemDraw es capaz de predecir espectros de H1 y C13, asignar picos y calcular las constantes de acoplamiento.

Después de aprender conceptos básicos sobre lectura de espectros, los estudiantes podrán realizar asignación de espectros “desconocidos”, de tal forma que podrán poner en marcha las ideas aprendidas gracias a la interfaz de ChemOffice.
Conclusiones
Desde isómeros hasta espectroscopía, ChemDraw hace posible la enseñanza de la química orgánica dentro de los centros de educación secundaria y avanzada. Gracias a esta interfaz los estudiantes podrán aumentar sus conocimientos químicos formándose de forma correcta para poder afrontar su futuro como científicos e investigadores.
- Detalles
- Categoría: ChemOffice
- Visto: 56194
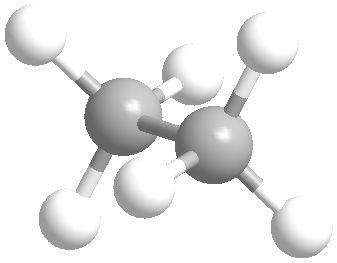
En esta noticia realizaremos una simulación sobre como dibujar proyecciones de Newman en ChemDraw, utilizaremos etano como ejemplo. Los puntos a seguir serán:
- Dibujar una molécula de metano
- Duplicaremos la estructura del metano y estableceremos una unión entre ambos
- Utilizaremos la herramienta “orbital” para dibujar el círculo de la proyección de Newman
- Rotaremos uno de los metanos sobre sí mismo 180˚
- Analizaremos por completo la proyección de Newman
Dibujo de una molécula de metano
Para dibujar una molécula de etano en proyección de Newman será preciso dibujar un metano en el frente y otro en el fondo, respectivamente. Para ello es tan sencillo como usar el comando “enlace sólido” para introducir el primer enlace simple y, a partir de ahí, dibujar la siguiente estructura:
")
Duplicado de una molécula de metano y establecimiento de una unión entre fragmentos

Seguidamente, podemos duplicar este fragmento de metano para generar la estructura del etano. Para ello deberemos seleccionar la molécula de metano previamente dibujada mediante la herramienta “selección” y arrastrar la figura para crear otra idéntica. A continuación, podemos unir los átomos de carbono centrales de cada metano para formar la molécula de etano.
Utilización de la herramienta “orbital” para dibujar el círculo de la proyección de Newman

Una vez listos los componentes para la proyección de Newman el siguiente paso será introducir en la estructura el círculo característico de este tipo de proyecciones. Para lo cual vamos a utilizar la herramienta “orbital” que podemos encontrar en View > Other Toolbars > Orbitals, o directamente en la columna izquierda de comandos de dibujo.
El tamaño del orbital está limitado al igual que el tamaño de los enlaces.
Rotación de uno de los fragmentos de metano sobre si mismo 180˚
Para rotar una de las mitades de metano será necesario seleccionarla previamente gracias a la herramienta “selección”.

A continuación seleccionamos Objects>Rotate>180º

El último paso será acercar uno de los fragmentos al otro, para conseguir de esa forma la proyección tipo Newman que buscábamos.

Si durante el dibujo de la proyección de Newman aparece en el centro del círculo una señal roja seleccione File > Preferences > des-seleccione la opción “Atom near other bonds”
Análisis completo de las proyecciones de Newman
Si analizamos los resultados de propiedades obtenidos mediante ChemDraw para las estructuras siguientes, puede apreciarse que para la interfaz todas y cada una de las estructuras son químicamente idénticas, pues sus puntos de ebullición y peso molecular son iguales. Lo mismo se aplicaría a cada una de las proyecciones de Newman que pudiéramos dibujar.

En el caso de realizar este tipo de análisis utilizando Chem3D, podemos simular optimizaciones energéticas para verificar cuál estructura presenta menor energía. Para ello abriremos el archivo de la molécula de etano en Chem3D y seleccionaremos los átomos que aparecen en la siguiente imagen para, a continuación, seleccionar Calculation > dihedral driver > single angle plot.

Aparecerá en la pantalla principal una gráfica donde aparece representada la energía respecto al ángulo diedro, donde podremos visualizar para qué ángulo la energía de la estructura será mínima siendo, en este caso, -60 y 60º.

Conclusión
ChemDraw es capaz de representar e interpretar proyecciones de Newman, además de las proyecciones de Fisher, Hanworth y las sistemáticas recomendadas por la IUPAC. Gracias a Chem3D podemos realizar estudios de optimización energética de las proyecciones de Newman dibujadas en ChemDraw, de esta forma conoceremos cuales son energéticamente más estables y, por tanto, cuales se encuentran en mayor abundancia de manera natural. Este tipo de estudios es muy útil para estructuras con más sustituyentes (no solo átomos de hidrógeno) donde se podrá estudiar la influencia del grupo sustituyente en la orientación del ángulo diedro.
- Detalles
- Categoría: Comsol
- Visto: 21585

Con estos cinco webinars de 30 minutos podrás seguir paso a paso la metodología de modelado multifísico de un transductor piezoeléctrico, utilizando COMSOL Multiphysics.
COMSOL Multiphysics es el software de modelado por elementos finitos más reconocido para la simulación de fenómenos multifísicos. Su auge en el sector es debido a su concepción desde los inicios de su desarrollo como herramienta multifísica, lo que le permite acoplar fenómenos físicos diferentes con naturalidad, desde una misma herramienta y con una metodología de trabajo homogénea y fácil de comprender.
Durante la semana multifísica, hemos realizado un curso de COMSOL Multiphysics on-line divido en cinco partes que se corresponden con las diferentes partes del flujo de trabajo para el modelado multifísico con COMSOL. Para ello hemos escogido un modelo fundamentalmente multifísico y fácil de entender como es un transductor piezoeléctrico que permite ver con cierta sencillez cómo se crea el modelo geométrico, se definen los materiales del transductor, se aplican y acoplan las diferentes físicas involucradas, se malla el modelo y se resuelve para posteriormente analizar los resultados en el postprocesado.
Sigue los enlaces a las grabaciones más abajo y fórmate como ingeniero de modelado multifísico con COMSOL Multiphysics.
- Detalles
- Categoría: Comsol
- Visto: 14295
Debido a su versatilidad inherente y a la respuesta frecuencial de banda ancha las antenas de ranura en espiral tienen una variedad de aplicaciones para diferentes bandas de frecuencias de microondas. Por ejemplo, estas antenas son utilizadas en comunicaciones inalámbricas, detección, posicionamiento y seguimiento. Para optimizar el diseño de las antenas de ranura en espiral, los ingenieros utilizan el análisis electromagnético para calcular con precisión características como los parámetros S y los diagramas de campo lejano.
Beneficios de las antenas de ranura en espiral
Las antenas de ranura en espiral tienen varias ventajas, entre las que se incluyen:
• Radiación polarizada circularmente prácticamente perfecta
• Respuesta frecuencial de banda ancha
• La capacidad de mantener un diagrama de radiación consistente e impedancia sobre un gran ancho de banda
Además, el diseño de las antenas de ranura en espiral permiten ser montadas adaptándose sobre una variedad de objetos. Esto es de gran utilidad, por ejemplo, en la industria de defensa, donde las antenas de ranura en espiral pueden montarse sobre automóviles y aviones militares y utilizarse para aplicaciones de comunicaciones y vigilancia.
Existen múltiples tipos de antenas en espiral, pero una de las más comunes es la antena espiral de Arquímedes. En la entrada del blog de COMSOL (enlace inferior), Caty Fairclough presenta los detalles para el modelado de esta antena con el software COMSOL Multiphysics® y el módulo RF Module.
- Detalles
- Categoría: Minitab
- Visto: 144599
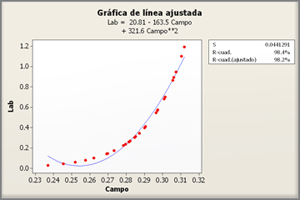
Por Jim Frost
En análisis de regresión, se desea que el modelo de regresión tenga variables significativas y obtener un valor R2 alto. Esta combinación bajo P valor/alto R2 indica que cambios en los predictores están relacionados con cambios en la variable de respuesta y que el modelo explica mucha de la variabilidad de la respuesta.
Esta combinación parece ir junta de forma natural. Pero ¿qué pasa si tu modelo de regresión tiene variables significativas pero explica poco la variabilidad? Tiene bajos valores P y bajos coeficientes de determinación R2.
A primera vista, esta combinación no tiene sentido. ¿Son los predictores de significancia todavía significativos? ¡Veamos esto!
Comparación de modelos de regresión con bajos y altos valores R2
Es difícil comprender esta situación utilizando únicamente números. La investigación muestra que los gráficos son esenciales para interpretar correctamente análisis de regresión. ¡La comprensión es más fácil cuando se puede ver lo que está pasando!
Con esto en mente, utilizaremos gráficos de líneas ajustadas. Sin embargo, un gráfico 2D de líneas ajustadas puede visualizar únicamente los resultados de una regresión simple, que tiene una variable predictiva y la respuesta. Los conceptos son ciertos para la regresión lineal múltiple, pero no se puede graficar las mayores dimensiones que se requieren.
Estos gráficos de líneas ajustadas muestran dos modelos de regresión que tienen ecuaciones de regresión prácticamente idénticas, pero el modelo superior tiene un bajo valor R2 mientras que el otro lo tiene alto. Se han mantenido las escalas de los gráficos constantes para una comparación más fácil. Aquí están los datos para estos ejemplos.
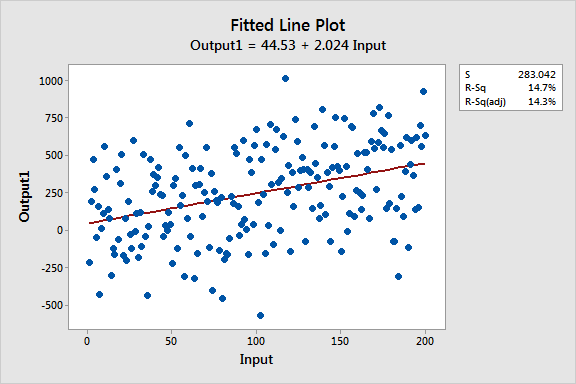
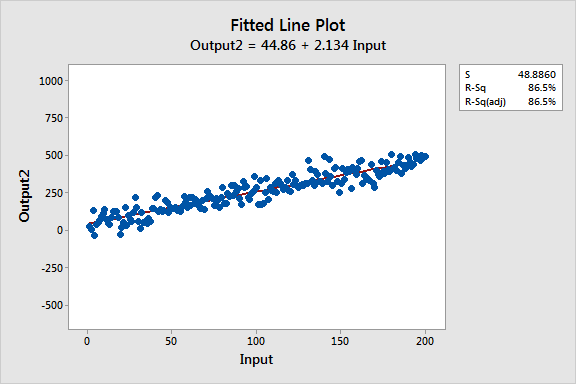
Similaridades entre los modelos de regresión
Los dos modelos son prácticamente idénticos en varios ámbitos:
- Ecuaciones de regresión: Salida = 44 + 2 * Entrada
- Entrada es significativa con P< 0.001 para ambos modelos
Se puede ver que la pendiente creciente de las dos líneas de regresión es aproximadamente 2, y siguen con precisión la tendencia que está presente en ambos conjuntos de datos.
La interpretación del valor P y el coeficiente para la Entrada no cambia. Si se mueve a la derecha en cualquier línea incrementando Entrada en una unidad, existe un incremento medio de dos unidades en Salida. Para ambos modelos, el valor P significativo indica que se puede rechazar la hipótesis nula que el coeficiente es igual a cero (sin efecto).
Más aún, si se entra el mismo valor para Entrada en ambas ecuaciones, calculará valores de predicción prácticamente equivalentes para Salida. Por ejemplo, una entrada de 10 produce una salida pronosticada de 66.2 para un modelo y 64.8 para el otro modelo.
Diferencias entre modelos de regresión
Seguro que la principal diferencia es lo que primero se ve de estos gráficos de líneas ajustadas: La variabilidad de los datos alrededor de dos líneas de regresión es drásticamente diferente. R2 y S (error estándar de la regresión) describen numéricamente esta variabilidad.
El gráfico de bajo R2 muestra que, aunque ruidosos, los datos de alta variabilidad pueden tener una tendencia significativa. La tendencia indica que la variable predictiva todavía proporciona información sobre la respuesta aunque los puntos de los datos caigan lejos de la línea de regresión. Mantengamos este gráfico en mente para cuando intentemos reconciliar variables significativas con un bajo valor R2!
Como vemos, las dos ecuaciones de regresión producen predicciones prácticamente idénticas. Sin embargo, los niveles de variabilidad diferentes afectan a la precisión de estas predicciones.
Para calcular la precisión, miraremos los intervalos de predicción. Un intervalo de predicción es un rango que es probable que contenga el valor de respuesta de una nueva observación única dados unos ajustes especificados de los predictores en el modelo. Los intervalos más estrechos indican predicciones más precisas. Debajo se encuentran los valores ajustados y los intervalos de predicción para una Entrada de 10.
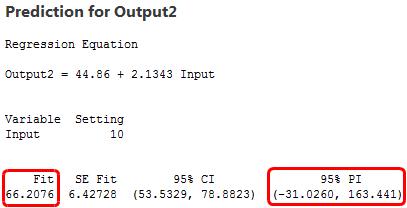
El modelo con los datos de alta variabilidad produce un intervalo de predicción que se extiende entre aproximadamente -500 a 630, ¡más de 1100 unidades! Mientras tanto, el modelo de baja variabilidad tiene un intervalo de predicción entre -30 y 160, aproximadamente 200 unidades. Claramente, las predicciones son mucho más precisas para el modelo de alto R2, aunque los valores ajustados sean prácticamente los mismos.
La diferencia en precisión debería tener sentido después de ver la variabilidad presente en los datos reales. Cuando los puntos de los datos se dispersan más, las predicciones deben de reflejar que añaden incerteza.
Reflexiones finales
Recapitulando lo que hemos aprendido:
- Los coeficientes estiman las tendencias mientras que R2 representa la dispersión alrededor de la línea de regresión.
- Las interpretaciones de las variables significativas son las mismas tanto para modelos con alto y bajo R2.
- Los valores R2 bajos son problemáticos cuando se necesitan predicciones precisas.
¿Entonces, qué hacer si se tienen predictores significativos pero una bajo valor R2? Alguno dirá, “añadir más variables al modelo!”
En algunos casos, es posible que los predictores adicionales puedan incrementar la potencia explicativa verdadera del modelo. Sin embargo, en otros casos, los datos contienen un cantidad inherentemente más alta de inexplicable variabilidad. Por ejemplo, muchos estudios psicológicos tienen valores R2 menores que 50% porque la gente es impredecible.
Las buenas noticias son que incluso cuando R2 sea bajo, los valores P bajos indican una relación real entre los predictores significativos y la variable respuesta.
- Detalles
- Categoría: ChemOffice
- Visto: 17708

La combinación de las funcionalidades de ChemDraw y ChemFinder permite crear bibliotecas combinatorias con predicción de propiedades. En esta noticia se abordarán las siguientes funciones:
- Expansión de estructuras genéricas en ChemDraw
- Exportación de una lista de estructuras expandidas a un SDFile
- Importación de SDFile en ChemFinder, uso como base de datos química
- Predicción de propiedades y asignación de nombres para cada estructura mediante ChemFinder
- Importación de SDFile en ChemDraw Excel
- Creación de estructuras genéricas
- Predecir propiedades y asignar nombres mediante ChemDraw Excel
Expansión de estructuras genéricas en ChemDraw
Comenzamos con una estructura genérica simple dibujada en ChemDraw, la cual expandimos para ver sus doce combinaciones.

Nuestro objetivo será crear una base de datos química con las doce sustancias, predecir la temperatura de fusión y de ebullición, por comparación directa. Se podrá, además, asignar el nombre a cada una de las doce sustancias. Usando la función “Expansión de estructura genérica” de ChemDraw, podemos visualizar las doce estructuras:

ChemDraw puede asignar puntos de fusión, ebullición y nombre sistemático de cada una de las estructuras, pero esto debería hacerse para cada sustancia, nuestro objetivo es asignar estas propiedades para todo el conjunto a la vez.
Exportación de una lista de estructuras expandidas a un SDFile
ChemDraw puede exportar múltiples estructuras en formato SDFile, formato estandarizado para estructuras químicas. Este SDFile puede importarse a ChemFinder o ChemDraw para Excel. El significado de exportar un archivo en SDFile supone un tipo de “guardado”, que puede usarse para convertir estructuras individuales en formato SKC, MOL y otros.

Importación de SDFile en ChemFinder, uso como base de datos química
A continuación, podemos abrir el archivo SDFile en ChemFinder para visualizar la base de datos química que hemos creado. Los pasos son:
- Archivo > Crear nueva base de datos
- Seleccionar > Formulario en blanco
- Archivo > importar > SDFile y seleccionamos el archivo previamente guardado
- Seleccionamos como base de datos de salida el valor predeterminadas
Por defecto, la pantalla de ChemFinder está configurada como se muestra a continuación

En la pantalla se mostrará la primera estructura de las doce que componen la base de datos, en nuestro caso, el fenol. A continuación, podemos asignar propiedades, función que permite ChemFinder Ultra.
Predicción de propiedades y asignación de nombres para cada estructura mediante ChemFinder
La opción “procesador de propiedades” de ChemFinder asigna propiedades a cada una de las sustancias de la base de datos, para ello será preciso seguir los siguientes pasos:
- Click derecho en un área en blanco de la ventana de visualización
- Seleccione “calcular propiedades” en el menú emergente
- Seleccione “punto de ebullición” en la sección del servidor ChemPropPro
- Seleccione “punto de congelación” en la selección del servidor ChemPropPro (al igual que en el punto anterior)
- Seleccione “nombre químico” en la selección del servidor CFW
- Seleccione la casilla de verificación “crear cuadros en el formulario para nuevos valores” (de esta forma los resultados se mostrarán en la pantalla)
- Haga click en “rellenar” para agregar los campos a la pantalla y a la base de datos

Con esto hemos creado una librería combinatoria en ChemFinder. Podemos visualizar las sustancias en la tabla de datos que se muestra a continuación. Los apodos “Me”, como en el caso del tolueno, se reemplazan por las estructuras reales, se conservarán en ChemDraw para Excel.

Crear una estructura genérica (con más combinaciones)
Vamos a realizar los mismos pasos para crear estructuras de mayor tamaño que las anteriores:
- El sustituyente hidrógeno es añadido a la lista inicial de combinaciones (aumenta a 13 el número de sustituyentes)
- Se añade un punto de “unión variable” a los otros cinco átomos de carbono del fenilo. ChemDraw es consciente de que solo hay tres puntos de unión químicamente diferentes, por lo que esta agregará 3 combinaciones
- Una lista de cuatro halógenos se añade a este punto de “unión variable” (4 combinaciones)
- Doce de estas estructuras, las que contienen el hidrógeno en la lista de sustituyentes, hacen que la posición del punto de “unión variable” sea químicamente equivalente
- Finalmente generamos las estructuras genéricas (13 · 3 · 4) – 12 = 148 estructuras
Importar las combinaciones de SDFile a ChemDraw para Excel
A continuación, importaremos el archivo SDFile en ChemDraw Excel. Una lista de pequeño tamaño puede ser importada en Excel, una lista de gran tamaño deberá ser importada en ChemFinder. Los pasos para importar dentro de ChemDraw Excel son:
- Abrimos Excel y creamos una nueva página en blanco
- Creamos una hoja de trabajo químicamente inteligente a través de ChemOffice > nueva hoja de trabajo ChemOffice
- Importar el archivo SDFile vía ChemOffice > Importar/exportar > Importar tabla
- Seleccionar el archivo que deseemos
Predicción de propiedades y asignación de nombres para cada una de las estructuras, usando ChemDraw para Excel
La predicción de nombre y propiedades puede añadirse como una función de Excel. Se agregan tres columnas con los siguientes encabezados y definiciones de funciones:
- Columna B, “nombre” = CFW_CHEMICAL_NAME(A2)
- Columna C, “BP” = CHEMPROPPRO_BOILING_POINT(A2)
- Columna D, “MP” = CHEMPROP_FREEZING(A2)
Estas son las funciones de Excel correspondientes a las funciones de ChemFinder que se comentaron en el apartado anterior.

Conclusiones
La opción de “expandir estructura” de ChemDraw puede usarse junto con la opción de importación/exportación de SDFile para crear librerías combinatorias de compuestos en ChemFinder o ChemDraw Excel. Este método es capaz de crear hasta 500 combinaciones de estructuras genéricas. Múltiples estructuras genéricas podrán ser exportadas desde ChemDraw a ChemFinder o ChemDraw para Excel.