- Detalles
- Categoría: Signals Notebook
- Visto: 32151

Mayo 2019
Esta versión de Signals Notebook agrega nuevas opciones en la configuración de tablas al permitir que los administradores definan sus propias tablas. También se brinda la posibilidad de definir campos de hipervínculo externos y de fecha en las plantillas del sistema, además de haber agregar un método alternativo de autonumeración mediante el cual los números son secuenciales a nivel mundial.
Las siguientes capacidades sólo están disponibles para usuarios y administradores de Signals Notebook Standard o Private Cloud y no están disponibles como parte de Signals Notebook Individual Edition.
Actualizaciones administrativas
Los administradores ahora pueden agregar campos de hipervínculo externos y de fecha a las plantillas del sistema. Al igual que otros campos, como los campos de atributo, estos se pueden establecer como Requeridos u Opcionales. Los campos son parte de los metadatos de un experimento o cuaderno.
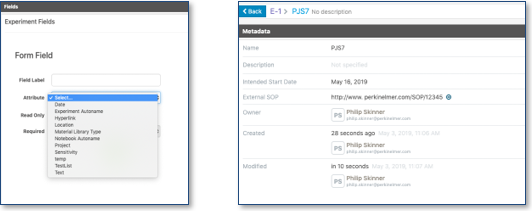
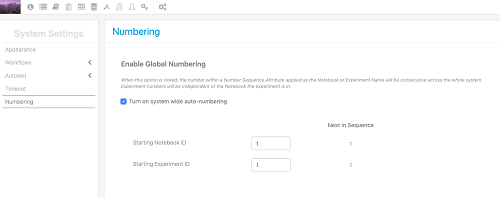
También se ha introducido una opción de numeración alternativa para permitir que la autonumeración ocurra secuencialmente en todo el sistema en lugar de dentro de un cuaderno. Con esta opción habilitada, el administrador puede avanzar la secuencia numérica pero no puede regresar a un número inferior. Esto proporciona un método para garantizar nombres de experimentos únicos.
Actualizaciones en las tablas
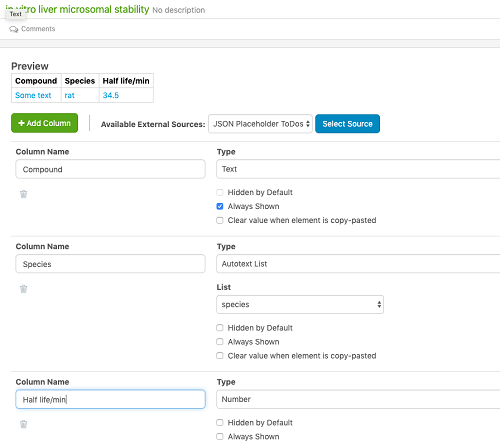
Los administradores de tablas ahora pueden definir las tablas que se agregarán a los experimentos. Cada tabla se define mediante una plantilla de tabla y el administrador puede definir los nombres de columna, los tipos de propiedad y si la columna está oculta de forma predeterminada, siempre se muestra, o si se elimina cualquier valor ingresado por el usuario cuando se copia un experimento. El administrador también puede definir compartir para controlar qué usuarios pueden agregar dichas tablas en sus experimentos.
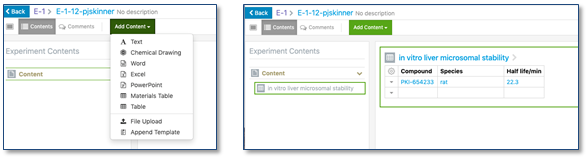
Una vez hecho esto el usuario podrá elegir dentro de cada experimento qué tabla quiere utilizar de entre la lista de tablas disponibles.
- Detalles
- Categoría: Comsol
- Visto: 5914

El límite de presentación avanzada de resúmenes para la Confrencia de COMSOL 2019 en Cambridge, es el próximo viernes 24 de mayo.
La presentación de sus trabajos en la conferencia es una gran oportunidad para compartir sus logros en simulación y modealdo con la comunidad de COMSOL. Envíe su resumen antes de este viernes y ahorre 120 euros en el registro a la conferencia.
Áreas temáticas sugeridas:
- Acústica
- Ingeniería química
- Electromagnetismo
- Flujo de fluidos
- Transferencia de calor
- Optimización
- Apps de simulación
- Mecánica estructural
- Detalles
- Categoría: Comsol
- Visto: 9185

Ya puede ver en nuestro canal de YouTube los vídeos de las semanas multifísicas 2018 y 2019.
En la semana de la multifísica 2018 ofrecimos el desarrollo de un modelo paso a paso en cinco sesiones de aproximadamente media hora, de forma que se podía seguir el modelado multifísico completo de un transductor Tonpilz.
Los vídeos describen las diferentes fases del flujo de trabajo característico de las simulaciones físicas: Creación de la geometría, asignación de materiales, ajuste de las ecuaciones físicas y condiciones de contorno, mallado, resolución numérica y postprocesado.
- Introducción y descripción del modelo. Inicialización. Creación de la geometría.
- Definiciones. Asignación de materiales.
- Ajuste de la Física, condiciones de contorno y acoplamientos.
- Mallado y resolución numérica.
- Postprocesado. Análisis y discusión de resultados.
En la semana de la multifísica 2019 ofrecimos el desarrollo de modelos de ejemplo para describir las principales características y funcionalidades incluidas en los módulos electromagnéticos de COMSOL.
Los módulos electromagnéticos de COMSOL permiten analizar y modelar dispositivos y sistemas electromagnéticos de diferentes frecuencias, desde dispositivos estáticos o de muy baja frecuencia, pasando por dispositivos que incluyen fenómenos de propagación de ondas de radiofrecuencias o microondas, hasta dispositivos ópticos, plasmas y semiconductores.
Consulte los vídeos sobre modelado electromagnético:
1. Herramientas del módulo de AC/DC.
2. Herramientas de los módulos RF y Óptica Ondulatoria.
3. Herramientas del módulo de Óptica de Rayos.
4. Herramientas del módulo de Plasmas.
5. Herramientas del módulo de Semiconductores.
Semana multifísica 2018: Desarrollo de un modelo paso a paso
Semana multifísica 2019: Módulos electromagnéticos de COMSOL Multiphysics
- Detalles
- Categoría: Minitab
- Visto: 6468

por Cody Steele
En esta época, no es raro que los errores en las entradas de datos se produzcan en conjuntos de datos tan grandes que no sea práctico buscar y corregir los errores a mano. Afortunadamente, Minitab LLCluye herramientas que facilitan la configuración de los datos, para que se puedan obtener las respuestas que se necesiten.
Digamos, por ejemplo, que se va a mirar en la base de datos Global Wood Density Database. Es una obra muy interesante si te conciernen las densidades de las maderas. Chave et al. lo llamaron "la mayor compilación de datos de densidad de madera hasta la fecha, que abarca 8412 taxones, 1683 géneros y 191 familias" (2009). Amablemente, sin embargo, la proporcionan en datadryad.org como un archivo Excel.
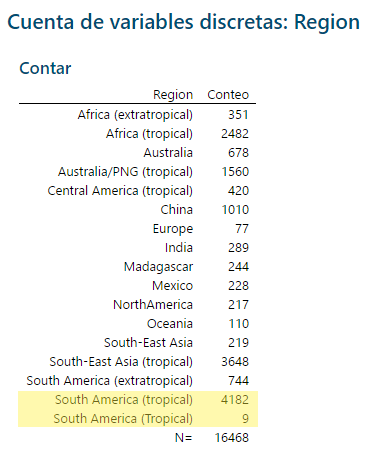
Pero resulta que hay un pequeño error en la columna Region (al menos en el momento de escribir este artículo). Es probable que apenas lo haya notado, pero hay una discrepancia entre mayúsculas y minúsculas. A un total de 4182 filas se les asigna la región de América del Sur (tropical), mientras que a 9 filas del conjunto de datos se les asigna la región de América del Sur (Tropical). Este es el tipo de desajuste que puede causar problemas en su análisis. Si sospecha que existe tal error, o simplemente quiere verificar que no existe, sería una tarea tediosa analizar 4191 filas de datos en busca de desajustes.
Afortunadamente, pueden encontrarse haciendo un recuento rápido en Minitab.
Encuéntralo
- Seleccione Estadísticas> Tablas> cuenta de variables individuales
- En Variables, ingrese Region. Haga clic en Aceptar.
En la tabla de salida, puede detectar la falta de coincidencia de mayúsculas/minúsculas en la parte inferior.
Arréglalo
Reparar los desajustes de mayúsculas es muye fácil en Minitab. Pruebe esto:
- Elija Datos> Recodificar> A texto.
- En Recodificar valores en las siguientes columnas, ingrese Region.
- En Método, seleccione Recodificar valores individuales.
- En la tabla que aparece, desplácese hacia abajo para encontrar la discrepancia entre mayúsculas y minúsculas. Luego, en la columna Valor recodificado, cambie South America (Tropical) para que use una t minúscula.
- En Ubicación de almacenamiento para las columnas recodificadas, seleccione En las columnas originales. Haga clic en Aceptar.
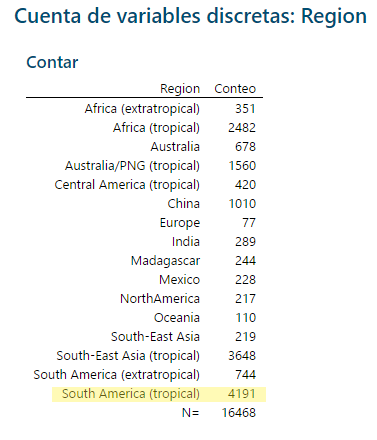
El resumen le mostrará las 9 instancias que fueron modificadas.

Arreglarlo antes de que sea un problema
Si está abriendo un archivo Excel, Minitab puede solucionar los desajustes entre mayúsculas y minúsculas incluso antes de que sepa que son un problema. Si tiene guardada la base de datos Global Wood Density Database y la abre, en Minitab, se le presentarán opciones para abrir un archivo Excel. Prueba esto:
- Elija Archivo>Abrir y seleccione el archivo Excel de su sistema de archivos.
- Haga clic en la pestaña titulada Data, el nombre de la hoja con los datos en el archivo original de Excel.
- Seleccionar Los datos tienen nombres de columna.
- Haga clic en Opciones.
- En Columnas de texto, seleccione Corregir desajustes de casos. Haga clic en Aceptar dos veces.
Si cuenta en la columna de regiones ahora, la corrección en la columna ya está hecha.
Envolver
Para obtener las respuestas necesarias de los datos, los datos deben estar lo suficientemente limpios para poder ser analizados. Minitab proporciona una serie de herramientas que se pueden utilizar para preparar los datos más rápidamente, y poder obtener información.
Referencias
Chave J, Coomes DA, Jansen S, Lewis SL, Swenson NG, Zanne AE (2009). Hacia un espectro mundial de la economía de la madera. Ecology Letters 12 (4): 351-366. http://dx.doi.org/10.1111/j.1461-0248.2009.01285.x
Zanne AE, Lopez-Gonzalez G, Coomes DA, Ilic J, Jansen S, Lewis SL, Miller RB, Swenson NG, Wiemann MC, Chave J (2009). Datos de: Hacia un espectro mundial de la economía de la madera. Repositorio Digital Dryad. http://dx.doi.org/10.5061/dryad.234
- Detalles
- Categoría: Comsol
- Visto: 6272

Informamos a los usuarios de COMSOL Multiphysics que ya está disponible COMSOL® 5.4 update 3. Esta actualización contiene mejoras de rendimiento y estabilidad para COMSOL Multiphysics®, COMSOL Server™, y COMSOL Client. La actualización puede aplicarse a las versiones de COMSOL® 5.4 (Build: 220 o 225), 5.4 update 1 (Build: 246), y 5.4 update 2 (Build: 295). La actualización puede aplicarse directamente a una instalación de la versión 5.4.
Si dispone de una versión anterior a la 5.4 y una licencia válida bajo suscripción del mantenimiento, entonces puede realizar una instalación completa de la versión 5.4 desde la página de descarga del producto, la que incluirá todas las actualizaciones.
La manera más sencilla de aplicar la actualización es arrancar el programa y seleccionar la opción Check for Product Updates (Buscar actualizaciones del producto). Si utiliza la versión del sistema operativo Windows®, ésta se localiza bajo el menú Archivo > Ayuda. Si utiliza Linux® o macOS, la encontrará en el menú de Ayuda.
- Detalles
- Categoría: BIOVIA
- Visto: 10309

La investigación en la industria de alimentos y bebidas busca impulsar la innovación de productos y respalda la propiedad intelectual y, por lo tanto, debe documentarse para respaldar las reclamaciones de productos, salvaguardar patentes, y conocimientos corporativos. Sin embargo, mientras que los científicos farmacéuticos han adoptado cuadernos de laboratorio electrónico (ELN) gracias a los cuales han encabezado su desarrollo, las empresas de alimentos y bebidas no eran (o son) muy partidarias de su uso. Recientemente una de estas empresas - Heinz - introdujo Biovia Notebook en sus herramientas de uso diario.
Retos
- Ciclos de vida de producto cortos que requieren innovación continua y rápida
- Capturar y compartir propiedad intelectual de manera eficiente
- Colaboración efectiva a través de espacios globales
Solución
- Cuaderno de laboratorio de Biovia, Biovia Notebook (anteriormente cuaderno de laboratorio de Accelrys, anteriormente Contur ELN)
Beneficios
- Mejora la eficiencia de investigación con protocolos estándar, así como la apariencia de los informes, permitiendo al usuario centrarse en la ciencia.
- Mejora del intercambio de conocimientos globales de forma instantánea gracias al IP de búsqueda, uso compartido, material reutilizable, etc.
- Implementación, despliegue global rápido y capacitación: ¡En menos de un año! Intercambio global de conocimientos y propiedad intelectual
La industria alimentaria, como sus hermanas las empresas de ciencias de la vida, también siente la necesidad de alejarse del uso del papel. Estas empresas son conscientes de los beneficios que supone el uso de cuadernos de laboratorio electrónico. Uno de los aspectos de mayor repercusión a la hora de tomar la decisión de utilizar o no un ELN es la posibilidad de compartir información de forma global con todos aquellos usuarios del mismo de forma segura y eficaz.
Cuando nos encontramos ante una empresa donde la producción es rápida (semanas o meses), frente a los tiempos de producción tan grandes que podemos encontrarnos en investigación farmacológica u otras empresas de ciencias de la vida, la necesidad de disponer de una organización eficaz crece exponencialmente, se requiere un acceso rápido a la información por parte de los trabajadores. Pero, hasta ahora, los ELN se habían mantenido fuera del alcance de la mayoría de las empresas de alimentos y bebidas, ya que las mismas veían estas herramientas más orientadas a empresas enfocadas a la ciencia de la vida y que, además, requerían de una gran infraestructura y grandes recursos para su puesta en marcha. En cambio, en la última década, estos ELN se han convertido en sistemas mucho más flexibles, que requieren pocos recursos para su instalación y uso y que, además, son fáciles de utilizar.
Brian Carman, Gerente y Jefe de Proyectos de I+D de Heinz, lo tiene claro - “Estamos felices de que las empresas farmacéuticas y de ciencias de la vida invirtieran en los ELN en el pasado, gracias a ello, han podido evolucionar hasta convertirse en una herramienta multidisciplinar, convirtiéndose en una mejor solución para nosotros”
El uso de ELN ha permitido en Heinz un aumento en la eficiencia, como afirma Brian Carman – “Cabría pensar que, por ser electrónico, cualquier ELN es mejor que el papel, pero algunos precisan de, al menos, 12 clics para permitir que el usuario participe en un experimento. Biovia Notebook te permite abrir cualquier entrada y, con un solo clic estás ahí, capturando ideas, tan fácil como pasar una página en un cuaderno de papel”.
Un aspecto que terminó de convencer a la compañía es la capacidad de realizar búsquedas dentro de sus experimentos para, así, ver cómo se había trabajado con anterioridad. Esto permite a los trabajadores beneficiarse de la sabiduría de sus compañeros, ayudando no solo a crear nuevos productos sino a solucionar problemas con los productos ya existentes.
En resumen, Biovia Notebook juega un papel clave en el desarrollo de productos de alta calidad en empresas alimentarias. “El acceso instantáneo a todos nuestros datos de investigación corporativa eliminan la redundancia y permiten a nuestros científicos mantenerse centrados”, concluyó Carman, de Heinz.
Más rentable, más rápido, más fácil.
- Detalles
- Categoría: Comsol
- Visto: 8081

COMSOL ha anunciado la disponibilidad del Cliente COMSOL para Android. Los ingenieros y estudiantes ahora pueden realizar tareas de simulación desde sus dispositivos Android, como teléfonos tablets y Chromebooks, simplemente conectándose con el software COMSOL Server que corre las simulaciones de forma remota.
COMSOL Client for Android amplía las capacidades de Application Builder y COMSOL Server permitiendo que los usuarios puedan utilizar aplicaciones de simulación desde cualquier sitio, sin estar limitados por el hardware del dispositivo. Al proporcionar COMSOL Multiphysics a los técnicos de campo o a los representantes de ventas, directamente en los dispositivos Android se permite que los usuarios lleven su trabajo de I+D in situ o como argumento de venta.
“COMSOL Server permite a los usuarios correr simulaciones a través de navegadores o clientes instalados en el escritorio,” explica Daniel Ericsson, jefe de productos de aplicaciones de COMSOL. “COMSOL Client for Android amplía las capacidades introduciendo una experiencia de usuario sin consturas sobre dispositivos Android."
Application Builder y COMSOL Server se desarrollaron para hacer que el modelado multifísico fuera accesible a una audiencia mucho más amplia. Application Builder permite a los especialistas de simulación crear aplicaciones hechas a medida basadas en sus propios modelos multifísicos. con COMSOL Server, las organizaciones ha podido distribuir herramientas de análisis específicas de su industria en un formato simplificado y rápido de implementar, acorde con COMSOL.
“Ampliar el alcance del modelado multifísico está en el núcleo de nuestra misión,” añade Ericsson. “COMSOL Client for Android permitirá a todo el mundo, desde ingenieros de diseño a técnicos de campo, ser más eficientes con la capacidad de correr simulaciones multifísicas desde la palma de sus manos.”
Las simulaciones se corrern en servidores remotos, por lo que los usuarios no están limitados por el hardware de su dispositivo. Los administratores continñuan teniendo completo control sobre quién puede acceder y correr a las aplicaciones utilizando COMSOL Server. Los usuarios de Android tendrán la última versión de una aplicación de simulación cada vez que abran la app.