
Del concepto a la implementación y el soporte (y todo lo demás): entendamos las 5 etapas de la FDA para la fabricación de dispositivos médicos
- Detalles
- Categoría: Minitab
- Visto: 3620
Por Ming-dong y coescrito por Abbie Wong, director de marketing de área de Minitab.
Cuando los efectos del COVID-19 causaron escasez de dispositivos médicos en todo el mundo, muchos fabricantes se apresuraron a modificar sus líneas de producción para satisfacer la demanda. Aunque la Food and Drug Administration de los Estados Unidos (FDA) relajó temporalmente las pautas para algunos productos como el desinfectante de manos, todavía existe una ruta de aprobación estándar los fabricantes de dispositivos médicos tienen que seguir para llevar sus productos al mercado.
Vamos a explorar los objetivos en cinco etapas y algunos ejemplos de las técnicas estadísticas que normalmente se llevan a cabo dentro de ellas.
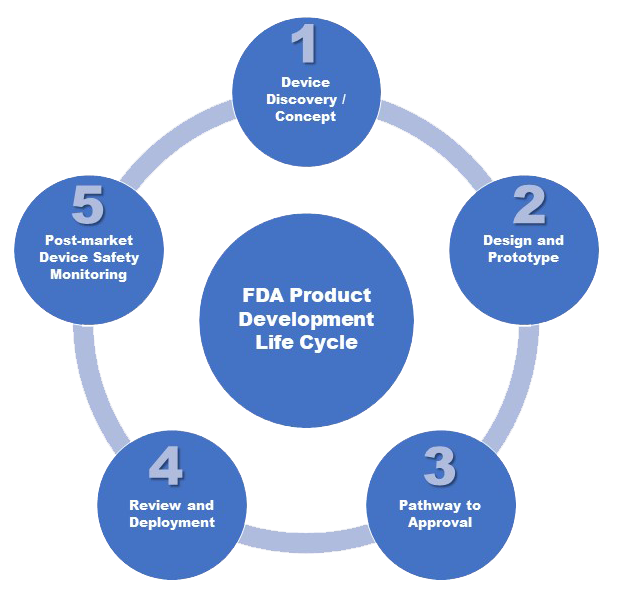
ETAPA 1: DESCUBRIMIENTO Y CONCEPTO DEL DISPOSITIVO
Probablemente habrá oído decir que la necesidad es la madre de la invención. Igual que ocurre con muchos productos nuevos y con la innovación en general, los fabricantes de dispositivos médicos a menudo inician un proyecto y avanzan a través de los objetivos de las etapas de la FDA porque ven una necesidad no satisfecha. Desde un marcapasos hasta una píldora con una cámara en su interior, todos los dispositivos médicos comienzan con una prueba de concepto, donde los ingenieros y técnicos de I+D verifican si el concepto es práctico.
A veces es posible que tengan algunos productos similares (o tal vez el mismo producto pero con diferentes materiales). Puede llevar sus datos sobre fiabilidad, consistencia y muchos otros factores a un Gráfico de variabilidad de Minitab que le ayudará a decidir cuál es el mejor producto para seguir adelante.
ETAPA 2: DISEÑO Y PROTOTIPO
En esta etapa, los investigadores construyen una versión previa del dispositivo médico, no para uso humano, sino para probar en entornos de laboratorio controlados. A medida que refinan el prototipo, continúan aprendiendo más sobre el uso potencial del producto para las personas y cómo reducir el riesgo de daño.
Ejemplo: predicción del ciclo de vida del producto mediante pruebas de vida aceleradas
Algunos productos, como los dispositivos médicos implantables, tienden a durar muchos años. Por esa razón, cada dispositivo médico debe estar etiquetado con una fecha de caducidad que esté respaldada por datos de vida útil.
Para recopilar datos de fallas, los investigadores pueden aplicar las de Minitab. ALT es cuando a un producto se le fuerza a fallar más rápidamente en condiciones extremas, como alta temperatura o alta presión. Llevar al producto para que falle más rápidamente reduce el tiempo de pruebas requerido. Luego, los datos de falla se analizan para extrapolar el ciclo de vida del producto en circunstancias normales.
ETAPA 3: CAMINO A LA APROBACIÓN
El camino hacia la aprobación de un dispositivo médico depende de su clasificación de riesgo (Clase I, Clase II o Clase III). A cada dispositivo se le asigna su clase según el nivel de control necesario para proporcionar una garantía razonable de su seguridad y eficacia. El sitio web de la FDA proporciona descripciones detalladas sobre cómo se clasifican los dispositivos y las pruebas y la validación requeridas para cada clase, pero para ilustrarlo de manera simple: la Clase I pueden ser dispositivos simples como vendas, guantes de látex y cepillos de dientes eléctricos, mientras que la Clase III suele ser de soporte vital, como los dispositivos médicos implantables del ejemplo anterior.
En esta etapa, se fabrica y prueba un lote de productos para ver su desempeño en relación con las especificaciones.
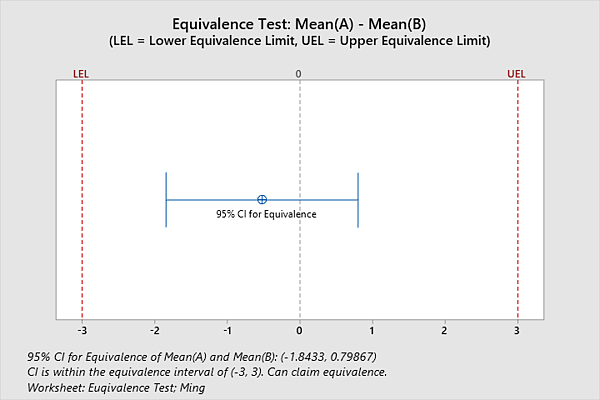
Ejemplo: Confirmación de que 2 dispositivos son equivalentes con prueba de equivalencia
Las pruebas de equivalencia estadística se pueden utilizar para evaluar si dos dispositivos médicos son equivalentes. La Prueba de equivalencia es un enfoque analítico para proporcionar evidencia de equivalencia. Al realizar una prueba de equivalencia, las hipótesis nula y alternativa tradicionales se invierten, lo que significa que la hipótesis nula es que dos dispositivos no son equivalentes (es decir, la diferencia entre ellos es enorme). La hipótesis alternativa es que son lo mismo. Por ejemplo, los investigadores quieren determinar si dos tipos de dispositivos de acceso intravenoso administran una cantidad equivalente de líquido. Definen la zona de equivalencia científica como una diferencia media en la cantidad de infusión de 3 ml o menos. A partir de la prueba de equivalencia de Minitab, los investigadores pueden estar seguros en un 95 % de que la diferencia en la cantidad media de infusión está entre -1,84334 y 0,798674 ml. Debido a que el intervalo de confianza del 95% se encuentra entre -3 y 3 (zona de equivalencia científica), los dos dispositivos intravenosos son equivalentes para las cantidades de infusión:

ETAPA 4: REVISIÓN E IMPLEMENTACIÓN DE LA FDA
En esta etapa, nos estamos preparando para presentar una solicitud ante la FDA para que podamos comercializar el dispositivo al público. Queremos demostrar que tenemos información suficiente y aceptable sobre la seguridad y eficacia del dispositivo. Supervisamos el proceso para asegurarnos de que sea estable y comprobamos que estamos fabricando el producto dentro de los límites de las especificaciones.
Una forma de mejorar un proceso de fabricación de dispositivos médicos es implementar un programa de control estadístico de procesos (SPC) . Normalmente utilizado en la producción en masa, un programa SPC permite a una empresa lanzar continuamente un producto utilizando gráficos de control, un gráfico de serie de tiempo especializado diseñado para ayudar a identificar patrones anormales de variabilidad en un proceso, en lugar de inspeccionar lotes individuales de un producto.
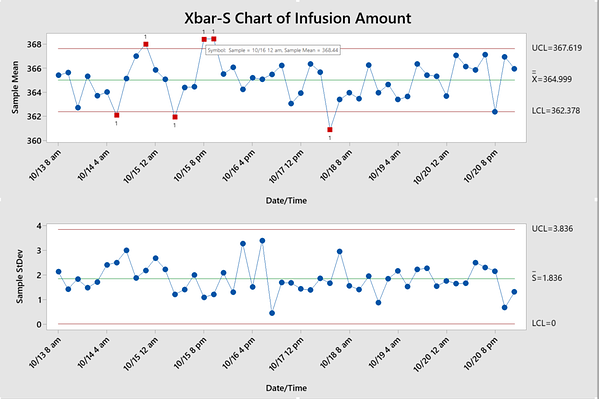
Ejemplo: Comprobación de cambios en las cantidades de infusión del dispositivo de acceso intravenoso
Se seleccionan al azar cinco dispositivos intravenosos de cada lote durante aproximadamente una semana. Se mide la cantidad de infusión. A partir de los gráficos de control a continuación, los ingenieros pueden identificar qué lotes están fuera de control. Al verificar los archivos de registro correspondientes, pueden identificar y eliminar toda variación de causa especial. Un programa SPC exitoso ayuda a los fabricantes a mantener procesos estables, mejorar la eficiencia y reducir costos, como se ve a continuación.
ETAPA 5: MONITORIZACIÓN DE SEGURIDAD POSTERIOR A LA COMERCIALIZACIÓN
Ahora que el producto ha sido lanzado, vamos a monitorizar la línea de producción adecuadamente para asegurarnos de que se mantenga adecuadamente y asegurar que el proceso permanezca en un estado de control constante. En esta etapa de vigilancia posterior a la comercialización, también nos aseguramos de que se informe y aborde cualquier evento adverso, como fallas o mal funcionamiento del dispositivo. La FDA lleva a cabo inspecciones de los fabricantes y emplea programas de informes que permiten a los fabricantes, profesionales de la salud y consumidores informar sobre problemas.
Ejemplo: diseño de un programa de mantenimiento preventivo utilizando análisis de confiabilidad
Esta etapa implica brindar un nivel de servicio adecuado y limitar el tiempo de inactividad de los dispositivos en la instalación. Una forma de mantenimiento es el , que es un evento programado. Los PM se programan de acuerdo con la clasificación de riesgo del dispositivo médico en diferentes momentos. Los estudios de vida útil de fiabilidad y el análisis de de Minitab ayudan a los fabricantes a calcular el riesgo de falla en un momento diferente del ciclo de vida para todo el producto y cada componente dentro del producto. Luego, los ingenieros pueden diseñar el programa de mantenimiento de acuerdo con la probabilidad de falla en las diferentes etapas del ciclo de vida.
TERMINANDO
Los fabricantes de dispositivos médicos deben asegurar la calidad y durabilidad en cada una de las cinco etapas del ciclo de vida del producto, y documentarlo detalladamente. Con suerte, estos ejemplos de la amplia gama de herramientas analíticas de Minitab pueden ayudarlo a comprender y avanzar en cada paso del ciclo de vida del producto.